The authors, probably mistakenly, considered their cases instances of Nievergelt syndrome (163400). Langer (1967) studied 2 cases and referred to the entity as 'mesomelic dwarfism of the hypoplastic ulna, fibula, mandible type.' ... A woman in this family with classic LWD was the mother of a fetus that inherited her deletion of SHOX; a second event, Turner syndrome, resulted in the absence of the SHOX allele on the paternal X chromosome. ... Both parents, who were heterozygous for the mutation, had Leri-Weill syndrome. In affected individuals with LWD or LMD from 12 Spanish multiplex families, 2 of which had previously been studied (Sabherwal et al. (2004, 2004)), Barca-Tierno et al. (2011) identified heterozygosity or homozygosity, respectively, for an A170P mutation (312865.0014) in the SHOX gene. ... Cytogenetics Fryns (1995) described a 15-year-old normally intelligent male who appeared to have both the blepharophimosis, ptosis, epicanthus inversus syndrome (BPES; 110100) and disproportionate dwarfism with marked shortening of the middle segments of the extremities with mandibular hypoplasia. ... Fryns (1995) suggested that the patient had a contiguous gene deletion syndrome, undetectable at the resolution of available cytogenetic techniques, involving 3q22.3-q23, where BPES is known to map, and also suggested that the locus for Langer mesomelic dwarfism is located in the same region.
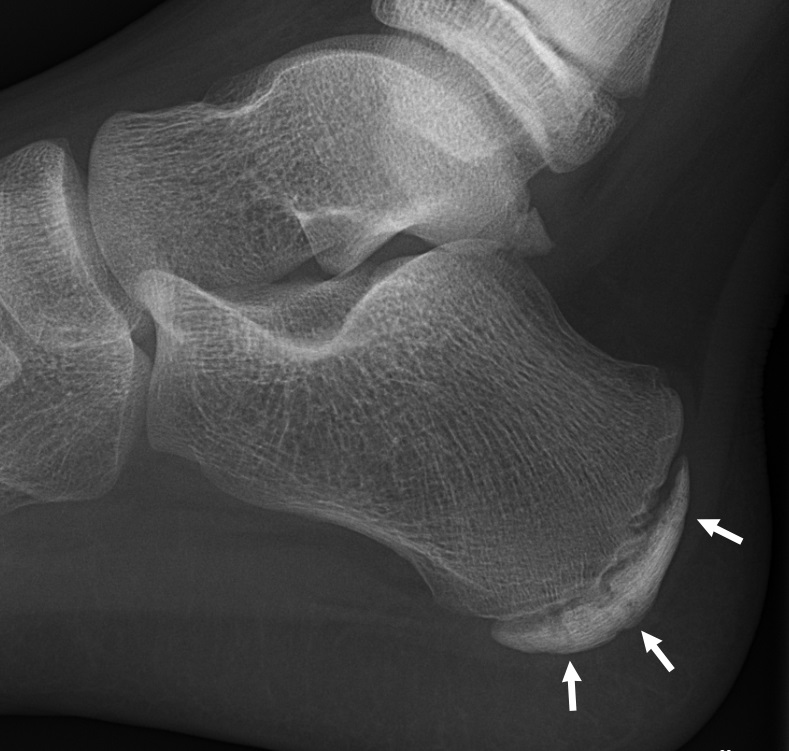
Langer mesomelic dysplasia is a disorder of bone growth. Affected individuals typically have extreme shortening of the long bones in the arms and legs (mesomelia). As a result of the shortened leg bones, people with Langer mesomelic dysplasia have very short stature. A bone in the forearm called the ulna and a bone in the lower leg called the fibula are often underdeveloped or absent, while other bones in the forearm (the radius ) and lower leg (the tibia) are unusually short, thick, and curved. Some people with Langer mesomelic dysplasia also have an abnormality of the wrist and forearm bones called Madelung deformity, which may cause pain and limit wrist movement. Additionally, some affected individuals have mild underdevelopment of the lower jaw bone (mandible).
This article has multiple issues. Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This article needs attention from an expert on the subject . Please add a reason or a talk parameter to this template to explain the issue with the article. When placing this tag, consider associating this request with a WikiProject . ( April 2014 ) This article may be too technical for most readers to understand . Please help improve it to make it understandable to non-experts , without removing the technical details. ( April 2014 ) ( Learn how and when to remove this template message ) This article's tone or style may not reflect the encyclopedic tone used on Wikipedia . See Wikipedia's guide to writing better articles for suggestions. ( April 2014 ) ( Learn how and when to remove this template message ) ( Learn how and when to remove this template message ) Langer mesomelic dysplasia Specialty Medical genetics Langer mesomelic dysplasia ( LMD ) is a rare congenital disorder characterized by an altered bone formation that causes a severe short and disproportionate stature.
A rare, genetic skeletal dysplasia characterized by severe disproportionate short stature with mesomelic and rhizomelic shortening of the upper and lower limbs. Epidemiology The exact prevalence is unknown. More than 100 cases have been described in the literature to date, with most of the patients being reported from populations with a high level of consanguinity. Clinical description Langer mesomelic dysplasia (LMD) is a more severe form of Léri-Weill dyschondrosteosis (LWD) and presents at birth with a severely shortened long bones of the limbs (involving both the middle and proximal segments), deformity of the humeral head, angulation of the radial shaft, carpal distortion, a short femoral neck, and absence or hypoplasia of the proximal half of the fibula. Mild hypoplasia of the mandible has been reported in some cases. In contrast to LWD, Madelung deformity is not typically present in LMD. Associated malformations are rare and intellect is normal in almost all reported LMD cases.
A number sign (#) is used with this entry because hyperprolinemia type II (HYRPRO2) is caused by homozygous or compound heterozygous mutation in the pyrroline-5-carboxylate dehydrogenase gene (P5CDH; 606811) on chromosome 1p36. For a discussion of genetic heterogeneity of hyperprolinemia, see HYRPRO1 (239500). Clinical Features Emery et al. (1968) described an affected mentally retarded 18-year-old girl whose retarded sister had died, presumably of the same disorder. Selkoe (1969) described a second type of hyperprolinemia with only mild mental retardation and without renal disease. Pavone et al. (1975) described 3 clinically normal sibs with type II hyperprolinemia.
Hyperprolinemia type 2 is an autosomal recessive proline metabolism disorder due to pyroline-5-carboxylate dehydrogenase deficiency. The condition is often benign but clinical signs may include seizures, intellectual deficit and mild developmental delay.
Hyperprolinemia type 2 results in an excess of a particular protein building block (amino acid), called proline, in the blood. This condition generally occurs when proline is not broken down properly by the body. Hyperprolinemia type 2 causes proline levels in the blood to be 10 to 15 times higher than normal, and it also causes high levels of a related compound called pyrroline-5-carboxylate. Some people with this condition develop mild intellectual disability and seizures; however, the symptoms of this disorder vary in severity among affected individuals.
Demand Avoidance is listed as a ‘sign or symptom of ASD’ (Appendix 3). [1] In response to the recent research Christopher Gillberg wrote a commentary article which stated “Experienced clinicians throughout child psychiatry, child neurology and paediatrics testify to its existence and the very major problems encountered when it comes to intervention and treatment.” [8] Proposed diagnostic criteria [ edit ] As of 2014 there are no recognized diagnostic criteria. [4] Proposed criteria by Newson include: Passive early history in the first year, avoiding ordinary demands and missing milestones Continuing to avoid demands, panic attacks if demands are escalated Surface sociability, but apparent lack of sense of social identity Lability of mood and impulsive Comfortable in role play and pretending Language delay , seemingly the result of passivity, often caught up quickly Obsessive behavior Neurological signs (awkwardness, similar to autism spectrum disorders [9] ) The underlying cause for this avoidance is said to be a high level of anxiety, usually from expectations of demands being placed on children, which can lead to a feeling of not being in control of a situation. [10] Children with PDA feel threatened when they are not in control of their environment and their actions, which triggers the fight, flight or freeze response. [11] History [ edit ] Newson first began to look at PDA as a specific syndrome in the 1980s when certain children referred to the Child Development Clinic at the University of Nottingham appeared to display and share many of the same characteristics. These children had often been referred because they seemed to show many autistic traits but were not typical in their presentation like those with classical autism or Asperger's syndrome . They had often been labelled as ' atypical autism ' or Persistent Development Disorder- Not Otherwise Specified (PDD-NOS). ... When Newson was made professor of developmental psychology at the University of Nottingham in 1994, she dedicated her inaugural lecture to talking about pathological demand avoidance syndrome. [12] In 1997, the PDA Society was established in the UK by parents of children with a PDA profile of autism. It became a registered charity in January 2016. [13] In July 2003, Newson published in Archives of Disease in Childhood for PDA to be recognized as a separate syndrome within the pervasive developmental disorders . [6] Bibliography [ edit ] Fidler, Ruth; Christie, Phil (2019). ... Journal of Child Psychology and Psychiatry . 55 (7): 758–768. doi : 10.1111/jcpp.12149 . ^ a b Newson E, Le Maréchal K, David C (July 2003). "Pathological demand avoidance syndrome: a necessary distinction within the pervasive developmental disorders" .
Histidinemia is a rare metabolic disorder characterized by elevated histidine levels in blood, urine, and cerebrospinal fluid, generally with no clinical repercussions. Epidemiology Reported prevalence varies widely from 1/8,600 to 1/180,000, probably as a result of differing screening programs. Clinical description Histidinemia is defined biochemically as elevated histidine in blood, urine and cerebrospinal fluid, and decreased levels of the metabolite urocanic acid in blood, urine, and the skin. In most individuals with histidinemia, the condition is clinically silent and considered benign, with no need for treatment or a specific diet. In a small subset of patients with specific events in the neonatal period, such as low oxygen, it has been suggested that histidinemia may contribute to development of intellectual disability, behavioral or learning disorders.
Histidinemia is an inherited condition characterized by elevated blood levels of the amino acid histidine, a building block of most proteins. Histidinemia is caused by the shortage (deficiency) of the enzyme that breaks down histidine. Histidinemia typically causes no health problems, and most people with elevated histidine levels are unaware that they have this condition. The combination of histidinemia and a medical complication during or soon after birth (such as a temporary lack of oxygen) might increase a person's chances of developing intellectual disability, behavioral problems, or learning disorders. Frequency Estimates of the incidence of histidinemia vary widely, ranging between 1 in 8,600 to 1 in 90,000 people.
Histidinemia is an inherited metabolic condition characterized by elevated levels of the amino acid histidine in blood, urine, and cerebrospinal fluid. In most cases, people with this condition have no health problems and may not even know that they are affected. Individuals with histidinemia who also experience a medical complication during or shortly after birth (such as a temporary lack of oxygen), may be at an increased risk of developing intellectual disability, behavioral problems, or learning disabilities. Histidinemia is caused by changes (mutations) in the HAL gene. This gene provides instructions for making an enzyme called histidase, which breaks down histidine into a molecule called urocanic acid. If histidase doesn't do its job properly, histidine levels become elevated.
A rare spotted fever rickettsiosis caused by infection with the tick-borne bacterium Rickettsia conorii , characterized by the onset of fever after an incubation period of about a week, followed by a centripetally spreading maculopapular rash, which may evolve into a petechial form. Accompanying symptoms are headaches, myalgia and/or arthralgia, among others. The typical ''tache noire'' may be observed at the site of the tick bite for several days. The disease is endemic in Africa, Southern Europe, and India.
2-methylbutyryl-CoA dehydrogenase deficiency is an organic acid disorder in which individuals lack adequate levels of an enzyme called 2-methylbutyryl-CoA dehydrogenase. This enzyme assists in the processing of a particular amino acid called isoleucine. The inability to process isoleucine correctly leads to the buildup of the amino acid in the body. The buildup can cause a variety of health problems, which vary widely from severe and life-threatening to mild or absent. Signs and symptoms of the disorder can begin a few days after birth or later in childhood.
A number sign (#) is used with this entry because short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) is caused by homozygous or compound heterozygous mutation in the gene encoding short/branched-chain acyl-CoA dehydrogenase (ACADSB; 600301). Description 2-Methylbutyryl-CoA dehydrogenase (MBD) deficiency is an autosomal recessive metabolic disorder of impaired isoleucine degradation. It is most often ascertained via newborn screening and is usually clinically asymptomatic, although some patients have been reported to have delayed development and neurologic signs. Therefore, the clinical relevance of the deficiency is unclear (Sass et al.., 2008). Clinical Features Andresen et al. (2000) reported a 3-year-old boy, born of consanguineous Pakistani parents, who showed increasing hypotonia and delayed motor development in the second year of life.
A rare organic aciduria characterized by impaired isoleucine degradation with increased plasma or whole blood C5 acylcarnitine levels (typically observed in newborn screening) and increased urinary excretion of N-methylbutyrylglycine. The condition is usually clinically asymptomatic, although patients with muscular hypotonia, developmental delay, and seizures (among others) have been reported.
Short/branched chain acyl-CoA dehydrogenase (SBCAD) deficiency (also known as 2-methylbutyryl-CoA dehydrogenase deficiency) is a rare disorder in which the body is unable to process proteins properly. Normally, the body breaks down proteins from food into smaller parts called amino acids. Amino acids can be further processed to provide energy for the body. People with SBCAD deficiency cannot process a particular amino acid called isoleucine. Most cases of SBCAD deficiency are detected shortly after birth by newborn screening, which identifies abnormal levels of certain compounds in the blood. In individuals with this condition, a compound called 2-methylbutyryl carnitine is elevated in the blood and another called 2-methylbutyrylglycine is elevated in the urine (2-methylbutyrylglycinuria).
A number sign (#) is used with this entry because of evidence that bone marrow failure syndrome-4 (BMFS4) is caused by homozygous mutation in the MYSM1 gene (612176) on chromosome 1p32. ... INHERITANCE - Autosomal recessive GROWTH Height - Short stature HEAD & NECK Head - Small head circumference (in some patients) Face - Dysmorphic facial features (in some patients) - Midface hypoplasia Ears - Deafness due to absence of the cochlear nerve (1 patient) - Low-set ears Mouth - Gingival hypertrophy Teeth - Delayed dentition RESPIRATORY - Recurrent respiratory infections CHEST External Features - Thoracic asymmetry SKELETAL Limbs - Rhizomelic limb shortening Hands - Short fingers SKIN, NAILS, & HAIR Skin - Dry skin - Eczema NEUROLOGIC Central Nervous System - Delayed development, mild (in some patients) HEMATOLOGY - Bone marrow failure - Anemia - Thrombocytopenia (in some patients) - Hypocellular bone marrow - Dysplastic red cell precursors - Features of myelodysplastic syndrome IMMUNOLOGY - Leukopenia - Decreased B cells - Decreased neutrophils - Hypogammaglobulinemia MISCELLANEOUS - Onset at birth - Three unrelated families have been reported (last curated September 2018) MOLECULAR BASIS - Caused by mutation in the Myb-like, SWIRM, and MPN domains-containing protein 1 gene (MYSM1, 612176.0001 ) ▲ Close
'Autism spectrum disorder,' sometimes referred to as ASD, is a broader phenotype encompassing the less severe disorders Asperger syndrome (see ASPG1; 608638) and pervasive developmental disorder, not otherwise specified (PDD-NOS). ... Mental retardation coexists in approximately two-thirds of individuals with ASD, except for Asperger syndrome, in which mental retardation is conspicuously absent (Jones et al., 2008). ... Mapping To study the genetics of autism, Alarcon et al. (2002) divided the syndrome into component autism-related traits (endophenotypes), hypothesizing that quantitative trait loci (QTLs) related to one or more of these traits might underlie putative or significant regions of autism linkage. ... Cytogenetics Poot et al. (2010) reported a boy with autism, delayed motor development, mild ataxia with poor coordination, hyperactivity, poor speech development, outbursts, and some features of Tourette syndrome (137580). The authors described a highly complex chromosomal rearrangement involving at least 3 breaks in chromosome 1 and 7 breaks in chromosome 7 on the paternally derived chromosome. ... Poot et al. (2010) suggested that haploinsufficiency for the CNTNAP2 gene may have caused the Tourette syndrome features, and that the combination of CNTNAP2 disruption and 1q41 deletion may have acted together to result in full-blown autism.
PIC is one of the so-called White Dot Syndromes . PIC has only been recognised as a distinct condition as recently as 1984 when Watzke identified 10 patients who appeared to make up a distinct group within the White Dot Syndromes . ... This is a complication, which can occur in other white dot syndromes and other eye conditions such as macular degeneration but occurs rarely in other forms of uveitis. ... Diagnosis [ edit ] Diagnosis of PIC can be difficult because the appearance may be similar to other conditions and types of posterior uveitis, especially other forms of the so-called white dot syndromes . The diagnosis is made by eliminating all the other possibilities by careful examination by an experienced ophthalmologist , aided with visual field testing and Fluorescein angiography (an intravenous dye used to show the blood vessels at the back of the eye). ... Review of Ophthalmology. 2004;11(6). ^ Polk TD, Goldman EJ. Chorioretinal Inflammatory Syndromes. International Ophthalmology Clinics. 1999;39(4):33-53.
Punctate inner choroidopathy (PIC) is an inflammatory disorder that primarily affects the choroid (vascular layer) of the eye. It most commonly occurs in young, near-sighted (myopic) women. The symptoms and severity may vary from person to person. Symptoms may include a blind spot ( scotomata ), blurred vision, photopsia (perceived flashes of light), floaters , light sensitivity ( photophobia ), distorted vision, and/or loss of peripheral vision. The majority of cases are self-limited (resolving on their own) with good visual prognosis, but permanent, severe visual loss can occur as a result of inflammation and complications such as growth of additional blood vessels ( neovascularization ) and scarring. The exact cause of PIC is not known, but it is thought to involve both genetic predisposition and environmental factors.
Delayed-Onset Form Granot et al. (1986) reported an Arab child in whom the diagnosis of partial CPS deficiency was first made when she presented at 9 years of age with hyperammonemic coma simulating Reye syndrome. Despite intensive therapy directed toward lowering of ammonia levels, she sustained irreversible brain damage.
A rare, severe disorder of urea cycle metabolism typically characterized by either a neonatal-onset of severe hyperammonemia that occurs few days after birth and manifests with lethargy, vomiting, hypothermia, seizures, coma and death or a presentation outside the newborn period at any age with (sometimes) milder symptoms of hyperammonemia. Epidemiology The worldwide prevalence ranges between 1/526,000-1,300,000 live births. Clinical description In the neonatal-onset form of carbamoyl-phosphate synthetase 1 deficiency (CPS1D), patients are usually healthy at birth but after few days they begin to manifest with lethargy and unwillingness to feed. Severe hyperammonemia continues and manifests with vomiting, hypothermia, hypotonia, seizures, coma, and can lead to death. Outside the newborn period, patients can present at any time in life.
Carbamoyl phosphate synthetase I deficiency (CPS1 deficiency) is a genetic disorder that causes episodes of toxic levels of ammonia in the blood (hyperammonemia). Symptoms include poor feeding, vomiting, lack of energy, low body temperature and weak muscle tone. These usually occur in the first few days of life. High levels of ammonia can lead to complications such as seizures, breathing problems, intellectual disability and coma. CPS1 deficiency is caused by alterations (mutations) in the CPS1 gene and is inherited in an autosomal recessive pattern. Diagnosis is based on symptoms, laboratory testing, and genetic testing.
Nomenclature Childhood-onset progressive bulbar palsy developing in the first and second decade of life was traditionally separated into Brown-Vialetto-Van Laere (BVVL) syndrome and Fazio-Londe syndrome (a BVVL-like syndrome without deafness). It is now known that Brown-Vialetto-Van Laere syndrome and Fazio-Londe syndrome are caused by biallelic pathogenic variants in either SLC52A2 or SLC52A3 . ... BVVL syndrome was first reported in 1894 by Charles Brown as an "infantile" form of ALS with associated hearing loss [Brown 1894]. ... Ernesto Vialetto in 1936 [Vialetto 1936] followed by the report of three sisters with similar clinical features by MJ Van Laere in 1966 resulted in the term BVVL syndrome being used to describe this pontobulbar palsy [Van Laere 1966]. Hereditary progressive bulbar palsy without deafness has also been described and referred to as Fazio-Londe syndrome [Fazio 1892, Londe 1894]; it was initially described in a mother and her son and later reported in two brothers with a more rapidly progressive syndrome.
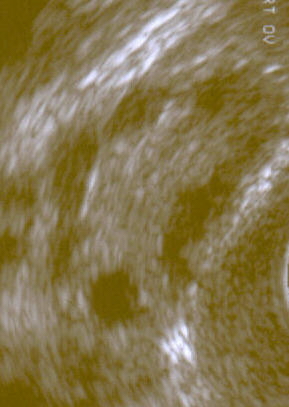
For the Colombian political party, see Comunitarian Party Option Seven . Polycystic ovary syndrome Other names Hyperandrogenic anovulation (HA), [1] polycystic ovarian disease, Stein–Leventhal syndrome [2] A polycystic ovary shown on an ultrasound image . ... Retrieved 13 March 2015 . ^ a b c d e f "Polycystic Ovary Syndrome (PCOS): Condition Information" . ... Retrieved 13 March 2015 . ^ a b c d Mortada R, Williams T (2015). "Metabolic Syndrome: Polycystic Ovary Syndrome". FP Essentials (Review). 435 : 30–42. ... "Nutritional management in women with polycystic ovary syndrome: A review study". Diabetes & Metabolic Syndrome (Review). 11 Suppl 1: S429–S432. doi : 10.1016/j.dsx.2017.03.030 . ... "A meta-analysis of polycystic ovary syndrome in women taking valproate for epilepsy".
Overview Gallstones are hardened deposits of digestive fluid that can form in your gallbladder. Your gallbladder is a small, pear-shaped organ on the right side of your abdomen, just beneath your liver. The gallbladder holds a digestive fluid called bile that's released into your small intestine. Gallstones Gallstones are hardened deposits of bile that can form in your gallbladder. Bile is a digestive fluid produced in your liver and stored in your gallbladder.
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome. Clinical Features Prasher et al. (1993) reported a 32-year-old man with mental retardation associated with an interstitial deletion of chromosome 2p13-p11.2 detected by G-banding studies.
Before organogenesis, the process is called embryo loss . [1] Resorption is more likely to happen early on in the gestation than later on; a later death of a fetus is likely to result in a miscarriage . [2] Contents 1 In rodents 2 In canines 3 See also 4 References 5 External links In rodents [ edit ] Fetal resorption in rats is common and can be influenced by antioxidants. [3] [4] [5] [6] [7] In canines [ edit ] In 1998, an ultrasound study found that the resorption of one or two conceptuses happen in up to 10% of all dog pregnancies, [2] although many cases of assumed complete resorption of an entire litter are likely to have just been the bitch experiencing a pseudopregnancy . [2] [8] See also [ edit ] Vanishing twin syndrome References [ edit ] ^ "Fetal Resorption - MeSH - NCBI" . www.ncbi.nlm.nih.gov . ^ a b c Edward C. ... External links [ edit ] MeSH C23.550.260.460.260 v t e Pathology of pregnancy , childbirth and the puerperium Pregnancy Pregnancy with abortive outcome Abortion Ectopic pregnancy Abdominal Cervical Interstitial Ovarian Heterotopic Embryo loss Fetal resorption Molar pregnancy Miscarriage Stillbirth Oedema , proteinuria and hypertensive disorders Gestational hypertension Pre-eclampsia HELLP syndrome Eclampsia Other, predominantly related to pregnancy Digestive system Acute fatty liver of pregnancy Gestational diabetes Hepatitis E Hyperemesis gravidarum Intrahepatic cholestasis of pregnancy Integumentary system / dermatoses of pregnancy Gestational pemphigoid Impetigo herpetiformis Intrahepatic cholestasis of pregnancy Linea nigra Prurigo gestationis Pruritic folliculitis of pregnancy Pruritic urticarial papules and plaques of pregnancy (PUPPP) Striae gravidarum Nervous system Chorea gravidarum Blood Gestational thrombocytopenia Pregnancy-induced hypercoagulability Maternal care related to the fetus and amniotic cavity amniotic fluid Oligohydramnios Polyhydramnios Braxton Hicks contractions chorion / amnion Amniotic band syndrome Chorioamnionitis Chorionic hematoma Monoamniotic twins Premature rupture of membranes Obstetrical bleeding Antepartum placenta Circumvallate placenta Monochorionic twins Placenta accreta Placenta praevia Placental abruption Twin-to-twin transfusion syndrome Labor Amniotic fluid embolism Cephalopelvic disproportion Dystocia Shoulder dystocia Fetal distress Locked twins Nuchal cord Obstetrical bleeding Postpartum Pain management during childbirth placenta Placenta accreta Preterm birth Postmature birth Umbilical cord prolapse Uterine inversion Uterine rupture Vasa praevia Puerperal Breastfeeding difficulties Low milk supply Cracked nipples Breast engorgement Childbirth-related posttraumatic stress disorder Diastasis symphysis pubis Postpartum bleeding Peripartum cardiomyopathy Postpartum depression Postpartum psychosis Postpartum thyroiditis Puerperal fever Puerperal mastitis Other Concomitant conditions Diabetes mellitus Systemic lupus erythematosus Thyroid disorders Maternal death Sexual activity during pregnancy Category