Walker–Warburg syndrome Other names HARD syndrome,Warburg syndrome Walker–Warburg syndrome has an autosomal recessive pattern of inheritance. Specialty Ophthalmology , neurology , medical genetics Walker–Warburg syndrome (WWS), also called Warburg syndrome , Chemke syndrome , HARD syndrome (Hydrocephalus, Agyria and Retinal Dysplasia), Pagon syndrome , cerebroocular dysgenesis (COD) or cerebroocular dysplasia-muscular dystrophy syndrome (COD-MD), [1] is a rare form of autosomal recessive congenital muscular dystrophy . [2] It is associated with brain ( lissencephaly , hydrocephalus , cerebellar malformations) and eye abnormalities. [3] This condition has a worldwide distribution. ... Almost all children with the disease die by the age of three. [8] Eponym [ edit ] WWS is named for Arthur Earl Walker and Mette Warburg (1926-2015), a Danish ophthalmologist. [9] [10] [11] Its alternative names include Chemke’s syndrome and Pagon’s syndrome, named after Juan M. ... Retrieved 2018-04-09 . ^ Reference, Genetics Home. "Walker-Warburg syndrome" . Genetics Home Reference . Retrieved 2018-04-17 . ^ Weiss, Thomas C.. "Walker-Warburg Syndrome - Facts and Information." Disabled World.
Cardiac involvement in FCMD/MEB occurs in the second decade of life in those who survive. FKTN-related Walker-Warburg syndrome is a more severe manifestation of the disorder, with death usually in the first year of life. ... Clinical Features This disorder has been described as FCMD/muscle-eye-brain disease (MEB) and the more severe Walker-Warburg syndrome; these designations have been retained here when used in the literature. ... Yoshioka and Kuroki (1994) performed clinical and genetic studies in 41 families with FCMD in Japan in an attempt to distinguish it from the Walker-Warburg syndrome and muscle-eye-brain disease, both of which, like FCMD, show an association of type II lissencephaly and ocular anomalies. ... Although the boy's parents were born in Henan and Shanxi Provinces and had no known Japanese ancestry, haplotype analysis showed that both mutant alleles were on Japanese-derived haplotypes. FKTN-Related Walker-Warburg Syndrome Silan et al. (2003) reported a Turkish patient with a severe congenital muscular dystrophy phenotype most closely resembling Walker-Warburg syndrome. ... Cotarelo et al. (2008) described a Spanish female infant, born of nonconsanguineous parents, who was diagnosed with Walker-Warburg syndrome and died on day 5 of life after suffering respiratory apnea and bradycardia.
The phenotype includes the alternative clinical designation Walker-Warburg syndrome (WWS). The disorder represents the most severe end of a phenotypic spectrum of similar disorders resulting from defective glycosylation of alpha-dystroglycan (DAG1; 128239), collectively known as 'dystroglycanopathies' (summary by Manzini et al., 2012). ... Clinical Features Manzini et al. (2012) reported 3 unrelated families with classic features of Walker-Warburg syndrome, including cobblestone lissencephaly, enlarged ventricles, cerebellar hypoplasia, eye abnormalities, and hypotonia. ... Inheritance The transmission pattern of Walker-Warburg syndrome in the families reported by Manzini et al. (2012) was consistent with autosomal recessive inheritance. Molecular Genetics In affected members of 3 unrelated consanguineous families with Walker-Warburg syndrome, Manzini et al. (2012) identified 3 different homozygous mutations in the GTDC2 gene (614828.0001-614828.0003).
The phenotype includes the alternative clinical designations Walker-Warburg syndrome (WWS) and muscle-eye-brain disease (MEB). ... Clinical Features Buysse et al. (2013) reported a family of East Indian descent in which 4 sibs had a clinical diagnosis of Walker-Warburg syndrome. Three pregnancies were terminated and 1 affected son died at 2 years of age.
Description Congenital muscular dystrophy-dystroglycanopathy with brain and eye anomalies (type A), which includes both the more severe Walker-Warburg syndrome (WWS) and the slightly less severe muscle-eye-brain disease (MEB), is an autosomal recessive disorder with characteristic brain and eye malformations, profound mental retardation, congenital muscular dystrophy, and death usually in the first years of life. ... All had a diagnosis of Walker-Warburg syndrome. Two of the patients were those reported by Chitayat et al. (1995) and Kanoff et al. (1998).
A number sign (#) is used with this entry because this form of congenital muscular dystrophy-dystroglycanopathy with brain and eye anomalies (type A5; MDDGA5), previously designated Walker-Warburg syndrome (WWS) or muscle-eye-brain disease (MEB), is caused by homozygous mutation in the FKRP gene (606596), which encodes a fukutin-related protein, on chromosome 19q13.3. ... Clinical Features Historically, the most severe forms of the dystroglycanopathies were described as Walker-Warburg syndrome (WWS) and muscle-eye-brain disease (MEB); these designations have been retained here when used in the literature. FKRP-Related Walker-Warburg Syndrome Cormand et al. (2001) reported a patient, born of consanguineous Pakistani parents, with WWS. ... Van Reeuwijk et al. (2010) reported a boy, born of consanguineous parents, with a clinical diagnosis of Walker-Warburg syndrome. He was born with severe hydrocephalus and showed limited spontaneous movements. ... In 2 sibs, the offspring of consanguineous parents, with a clinical diagnosis of Walker-Warburg syndrome, van Reeuwijk et al. (2010) identified a homozygous mutation in the FKRP gene (M1V; 606596.0019).
A number sign (#) is used with this entry because this form of congenital muscular dystrophy-dystroglycanopathy with brain and eye anomalies (type A3; MDDGA3), previously designated Walker-Warburg syndrome (WWS) or muscle-eye-brain disease (MEB), is caused by homozygous or compound heterozygous mutation in the POMGNT1 gene (606822) on chromosome 1p34. ... Clinical Features Historically, the most severe forms of the dystroglycanopathies were described as Walker-Warburg syndrome (WWS) and muscle-eye-brain disease (MEB); these designations have been retained here when used in the literature. ... Muscle-eye-brain disease has phenotypic similarities with the Walker-Warburg syndrome, another congenital muscular dystrophy caused by hypoglycosylation of alpha-dystroglycan (DAG1). ... Seven patients and 8 fetuses from 8 families were classified as having Walker-Warburg syndrome by a more severe presentation, including hydrocephalus, severe cobblestone complex, encephalocele, absent corpus callosum, and eye abnormalities. ... Taniguchi et al. (2003) identified 7 disease-causing mutations in the POMGNT1 gene among 6 non-Finnish Caucasian, Japanese, and Korean patients with suspected MEB, severe FCMD (MDDGA4; 253800), or Walker-Warburg syndrome. Mutations were dispersed throughout the entire POMGNT1 gene.
Early Descriptions of Walker-Warburg Syndrome The first case of Walker-Warburg syndrome is said to be that reported by Walker (1942) and labeled lissencephaly (Greek, 'smooth brain'). ... They termed the entity cerebro-ocular dysplasia/muscular dystrophy (COD-MD) syndrome. Heggie et al. (1987) reported a brother and sister with COD-MD syndrome, each of whom died at the age of about 1 year. ... Heggie et al. (1987) suggested that COD-MD syndrome may be identical to Walker-Warburg syndrome. Dobyns et al. (1989) also suggested that Walker-Warburg syndrome is similar to, if not identical to, cerebrooculomuscular syndrome (Heggie et al., 1987, Korinthenberg et al., 1984). ... Kim et al. (2004) reported a Japanese boy with Walker-Warburg syndrome. Prenatal studies showed a meningoencephalocele.
A number sign (#) is used with this entry because this form of congenital muscular dystrophy-dystroglycanopathy with brain and eye anomalies (type A6; MDDGA6), previously designated Walker-Warburg syndrome (WWS) or muscle-eye-brain disease (MEB), is caused by homozygous or compound heterozygous mutation in the LARGE gene (603590) on chromosome 22q12. ... Description Congenital muscular dystrophy-dystroglycanopathy with brain and eye anomalies (type A), which includes both the more severe Walker-Warburg syndrome (WWS) and the slightly less severe muscle-eye-brain disease (MEB), is an autosomal recessive disorder with characteristic brain and eye malformations, profound mental retardation, congenital muscular dystrophy, and death usually in the first years of life. ... Clinical Features Historically, the most severe forms of the dystroglycanopathies were described as Walker-Warburg syndrome (WWS) and muscle-eye-brain disease (MEB); these designations have been retained here when used in the literature. LARGE-Related Walker-Warburg Syndrome Van Reeuwijk et al. (2007) reported 2 Saudi sibs, born of consanguineous parents, with Walker-Warburg syndrome. ... The phenotype was consistent with Walker-Warburg syndrome. The patient had head control, but also had increased serum creatine kinase, absent alpha-dystroglycan on muscle biopsy, and mental retardation.
The phenotype includes the alternative clinical designations Walker-Warburg syndrome (WWS) and muscle-eye-brain disease (MEB). ... One had progressive microcephaly, and the other developed scoliosis and hypoventilation syndrome. Von Renesse et al. (2014) noted that the phenotype in their patients differed somewhat from that reported by Jae et al. (2013).
A number sign (#) is used with this entry because this form of congenital muscular dystrophy-dystroglycanopathy with brain and eye anomalies (type A2, MDDGA2), previously designated Walker-Warburg syndrome (WWS) or muscle-eye-brain disease (MEB), is caused by homozygous or compound heterozygous mutation in the POMT2 gene (607439) on chromosome 14q24.3. ... Description Congenital muscular dystrophy-dystroglycanopathy with brain and eye anomalies (type A), which includes both the more severe Walker-Warburg syndrome (WWS) and the slightly less severe muscle-eye-brain disease (MEB), is an autosomal recessive disorder with characteristic brain and eye malformations, profound mental retardation, congenital muscular dystrophy, and death usually in the first years of life. ... Clinical Features Historically, the most severe forms of the dystroglycanopathies were described as Walker-Warburg syndrome (WWS) and muscle-eye-brain disease (MEB); these designations have been retained here when used in the literature. POMT2-Related Walker-Warburg Syndrome Van Reeuwijk et al. (2005) reported 3 unrelated patients with POMT2-related WWS. ... As part of a larger study involving 92 probands with muscular dystrophy and evidence of a dystroglycanopathy, Godfrey et al. (2007) defined MEB as congenital muscular dystrophy with brain abnormalities less severe than that seen in Walker-Warburg syndrome. MRI findings in MEB included pachygyria with preferential frontoparietal involvement, polymicrogyria, cerebellar hypoplasia, cerebellar dysplasia, and frequent flattening of the pons and brainstem.
A number sign (#) is used with this entry because this form of limb- girdle muscular dystrophy-dystroglycanopathy (type C8; MDDGC8), also known as LGMDR24, is caused by homozygous or compound heterozygous mutation in the POMGNT2 gene (614828) on chromosome 3p22. Biallelic mutation in the POMGNT2 gene can also cause congenital muscular dystrophy-dystroglycanopathy with brain and eye anomalies (type A8; MDDGA8; 614830), a much more severe disorder. Description MDDGC8 is an autosomal recessive muscular dystrophy with onset in childhood. The phenotype is highly variable: some patients may have gait difficulties and impaired intellectual development, whereas others may be clinically asymptomatic. Common features include calf hypertrophy and increased serum creatine kinase, and muscle biopsy often shows dystrophic features (summary by Endo et al., 2015).
Heterotaxy syndrome is a condition in which the internal organs are abnormally arranged in the chest and abdomen. ... Unlike situs inversus, the abnormal arrangement of organs in heterotaxy syndrome often causes serious health problems. ... Frequency The prevalence of heterotaxy syndrome is estimated to be 1 in 10,000 people worldwide. ... Some people with heterotaxy syndrome have no identified gene mutations or other risk factors. ... However, about 10 percent of people with heterotaxy syndrome have a close relative (such as a parent or sibling) who has a congenital heart defect without other apparent features of heterotaxy syndrome.
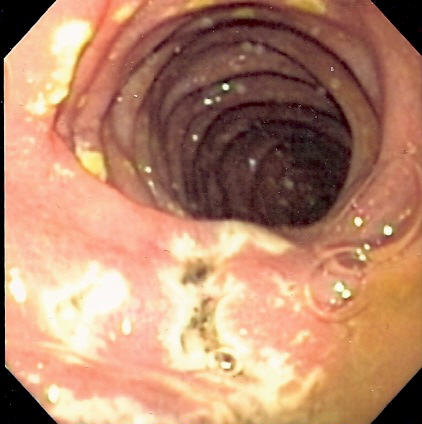
Condition in which tumours stimulate excessive gastric acid production Zollinger–Ellison syndrome Other names gastrinoma, pancreatic ulcerogenic tumor syndrome, ZES, Z-E syndrome [1] Endoscopy image of multiple small ulcers in the distal duodenum in a patient with Zollinger–Ellison syndrome Specialty Endocrinology Causes Gastrinoma Zollinger–Ellison syndrome ( Z-E syndrome ) is a disease in which tumors cause the stomach to produce too much acid, resulting in peptic ulcers . ... The American Surgical Association meeting in Philadelphia in April 1955 heard the first public description of the syndrome, and Zollinger and Ellison subsequently published their findings in Annals of Surgery . [17] References [ edit ] ^ "Zollinger Ellison syndrome" . ... PMID 20833329 . ^ Cho MS, Kasi A. Zollinger Ellison Syndrome. [Updated 2020 Jun 29]. In: StatPearls [Internet]. ... PMID 2565843 . ^ "Zollinger-Ellison Syndrome. Information about ZES Syndrome" . ... Retrieved 2018-04-17 . ^ "Zollinger Ellison Syndrome - NORD (National Organization for Rare Disorders)" .
Symptoms Symptoms of Zollinger-Ellison syndrome may include: Stomach pain. Diarrhea. ... Causes The exact cause of Zollinger-Ellison syndrome isn't known. But the pattern of events that occurs in Zollinger-Ellison syndrome typically follows the same sequence. The syndrome begins when one or more tumors form in your pancreas or a part of your small intestine called the duodenum. ... What treatments are available for Zollinger-Ellison syndrome, and which do you recommend for me? ... Are there websites you recommend to learn more about Zollinger-Ellison syndrome? Are any other medical problems more likely to occur because I have Zollinger-Ellison syndrome?
Impairments primarily associated with cannabis use Amotivational syndrome Specialty Psychology Amotivational syndrome is a chronic psychiatric disorder characterized by signs that are linked to cognitive and emotional states such as detachment , blunted emotion and drives, executive functions like memory and attention , [1] disinterest, passivity, apathy, and a general lack of motivation . [2] [3] This syndrome can be branched into two subtypes - marijuana amotivational syndrome, interchangeably known as cannabis induced amotivational syndrome which is caused by usage and/or dependency of the substance and is primarily associated with long-term effects of cannabis use, [1] and SSRI-induced amotivational syndrome or SSRI-induced apathy caused by the intake of SSRI medication dosage. [4] [5] According to the Handbook of Clinical Psychopharmacology for Therapists, amotivational syndrome is listed as a possible side effect of SSRIs in the treatment of clinical depression. [6] Contents 1 Signs & Symptoms 2 Subtypes 2.1 Cannabis Amotivational Syndrome 2.2 SSRI-Induced Amotivational Syndrome 3 Treatment & Measurement 3.1 Cannabis Amotivational Syndrome 3.2 SSRI-Induced Amotivational Syndrome 4 Current Research & Discourse 4.1 Cannabis Amotivational Syndrome 4.2 SSRI-Induced Amotivational Syndrome 5 See also 6 References Signs & Symptoms [ edit ] Amotivational syndrome has been suspected to affect the frontal cortex or frontal lobe of the brain by the impairment of that region [7] which monitors cognitive functions and skills that revolve around emotional expression , decision making , prioritisation , and internal, purposeful mental action. ... Subtypes [ edit ] Cannabis Amotivational Syndrome [ edit ] Legal cannabis (marijuana) product. Overconsumption and reliance could lead to cannabis-induced amotivational syndrome. The term amotivational syndrome was first devised to understand and explain the diminished drive and desire to work or compete among the population of youth who are frequent consumers of cannabis and has since been researched through various methodological studies with this focus on cannabis, or marijuana. [4] Cannabis amotivational syndrome is often used interchangeably with marijuana amotivational syndrome and marijuana or cannabis induced or related amotivational syndrome. ... Amotivational syndrome caused or related to SSRI dosage is also commonly known as apathy syndrome, SSRI-induced apathy syndrome, SSRI-induced apathy, and antidepressant apathy syndrome. . ... "Negative dimension in psychiatry. Amotivational syndrome as a paradigm of negative symptoms in substance abuse".
Milk-alkali syndrome Other names Calcium-alkali syndrome [1] In medicine , milk-alkali syndrome is characterized by high blood calcium and metabolic alkalosis caused by taking in too much calcium and absorbable alkali ; common sources of calcium and alkali are dietary supplements taken to prevent osteoporosis and antacids . ... Impaired kidney function is a risk factor but even people with healthy kidneys can develop the syndrome. [5] Diagnosis [ edit ] To diagnose milk-alkali syndrome, primary hyperparathyroidism has to be excluded. ... "Health-behavior induced disease: return of the milk-alkali syndrome" . J Gen Intern Med . 22 (7): 1053–5. doi : 10.1007/s11606-007-0226-0 . ... PMID 17483976 . ^ Beall DP, Henslee HB, Webb HR, Scofield RH (May 2006). "Milk-alkali syndrome: a historical review and description of the modern version of the syndrome". ... Last updated Update Date: 7 November 2013 by:Brent Wisse. Medline Plus: Milk-alkali syndrome ^ a b c Scofield RH et al. for eMedicine.
Intellectual disability, seizures, and other health problems can also occur in people with MVA syndrome. There are at least three types of MVA syndrome, each with a different genetic cause. ... Some people with MVA syndrome type 2 have unusually short arms. ... Individuals with this form may also have other signs and symptoms typical of MVA syndrome type 1. Frequency MVA syndrome is a rare condition. ... Causes BUB1B gene mutations cause MVA syndrome type 1, CEP57 gene mutations cause MVA syndrome type 2, and TRIP13 gene mutations cause the other form of MVA syndrome. Some people with MVA syndrome do not have mutations in any of these genes.
Jacquemont et al. (2002) stated that 14 cases of MVA syndrome had been reported during the previous 10 years. ... They described a boy in whom MVA syndrome was associated with myelodysplasia with a monosomy 7 bone marrow clone (252270). ... Callier et al. (2005) tabulated the clinical and cytogenetic findings in 28 reported cases of MVA syndrome. They also reported the first case of MVA syndrome without microcephaly and suggested that microcephaly is not mandatory for the diagnosis of the disorder. ... However, the patient had no other features of the mosaic variegated aneuploidy syndrome, such as poor growth, microcephaly, or mental retardation. ... The report expanded the phenotype associated with BUB1B mutations and the mosaic variegated aneuploidy syndrome to include common adult-onset cancers.
A number sign (#) is used with this entry because mosaic variegated aneuploidy syndrome-2 (MVA2) is caused by homozygous or compound heterozygous mutation in the CEP57 gene (607951) on chromosome 11q21. Description Mosaic variegated aneuploidy syndrome is an autosomal recessive disorder characterized by poor growth and variable phenotypic manifestations, such as facial dysmorphism and congenital heart defects, associated with mosaic aneuploidies resulting from defects in cell division (summary by Snape et al., 2011).
A number sign (#) is used with this entry because of evidence that mosaic variegated aneuploidy syndrome-3 (MVA3; 617598) is caused by homozygous mutation in the TRIP13 gene (604507) on chromosome 5p15.
Mosaic variegated aneuploidy (MVA) syndrome is a very rare condition characterized by problems with cell division (specifically during mitosis) that results in a high number of cells with missing (monosomy) or extra (trisomy) genetic material in multiple chromosomes and tissues (mosaic aneuploidies). ... The risk for cancer is increased, with rhabdomyosarcoma , Wilm's tumor , and leukemia reported in several cases. MVA syndrome is an autosomal recessive condition.
Mosaic variegated aneuploidy (MVA) syndrome is a chromosomal anomaly characterized by multiple mosaic aneuploidies that leads to a variety of phenotypic abnormalities and cancer predisposition. ... Differential diagnosis Aneuploidy can be a feature of chromosomal instability syndromes including Roberts syndrome, ataxia telangiectasia, xeroderma pigmentosum, Bloom syndrome, Werner syndrome and Nijmegen breakage syndrome. ... Cases with a cytogenetic confirmation of MVA syndrome and/or demonstrated BUB1B mutations should be offered Wilms tumor surveillance with renal ultrasonography every three to four months until five years.
Structure of dermatan sulfate, one of the molecules that accumulates in the lysosomes of MPS I patients Causes Deficiency of the alpha-L iduronidase enzyme Differential diagnosis Hunter syndrome ; other mucopolysaccharidoses Treatment Enzyme replacement therapy with iduronidase ; surgery Prognosis Death usually occurs before 12 years (Hurler syndrome/severe form); lifespan may be normal (Scheie syndrome/attenuated form) Frequency 1:100,000 (Hurler syndrome/severe); 1:115,000 (Hurler-Scheie syndrome/intermediate); 1:500,000 (Scheie syndrome/attenuated) [1] Mucopolysaccharidosis type I is a spectrum of diseases in the mucopolysaccharidosis family. ... Death usually occurs by age 10. [2] [3] In less severe cases, (Scheie syndrome, or attenuated MPS I), the presentation can vary considerably. ... The other two types are MPS I S ( Scheie syndrome ) and MPS I H-S ( Hurler-Scheie syndrome ). [ citation needed ] Because of the substantial overlap between Hurler syndrome, Hurler-Scheie syndrome, and Scheie syndrome, some sources consider these terms to be outdated. ... Retrieved 11 May 2018 . ^ Banikazemi, Maryam (12 Oct 2014). "Hurler syndrome, Hurler-Scheie Syndrome, and Scheie Syndrome (Mucopolysaccharidosis Type I)" . ... Retrieved 10 May 2018 . v t e Lysosomal storage diseases : Inborn errors of carbohydrate metabolism ( Mucopolysaccharidoses ) Catabolism MPS I Hurler Syndrome , Hurler-Scheie Syndrome , Scheie Syndrome MPS II: Hunter Syndrome MPS III: Sanfilippo Syndrome MPS IV: Morquio Syndrome MPS VI: Maroteaux-Lamy Syndrome MPS VII: Sly Syndrome MPS IX: Hyaluronidase deficiency
While affected individuals have traditionally been classified as having one of three MPS I syndromes (Hurler syndrome, Hurler-Scheie syndrome, or Scheie syndrome), no easily measurable biochemical differences have been identified and the clinical findings overlap; thus, affected individuals are best described as having either severe or attenuated MPS I, a distinction that influences therapeutic options. ... While affected individuals have traditionally been classified as having one of three MPS I syndromes (Hurler syndrome, Hurler-Scheie syndrome, or Scheie syndrome), no easily measurable biochemical differences have been identified [Muenzer 2004] and the clinical findings overlap; thus, affected individuals are best described as having either severe or attenuated MPS I, a distinction that influences therapeutic options. ... Data from the international MPS I registry available in 2011 showed that of the 891 individuals included in the registry 57% were classified as having Hurler syndrome, 23.5% as Hurler-Scheie syndrome, and 10% as Scheie syndrome; 8.6% were classified as either unknown or indeterminate. ... Attenuated MPS I (Hurler-Scheie Syndrome / Scheie Syndrome) If development is normal at age 24 months and if moderate somatic involvement is evident, an individual should be classified as having attenuated MPS I. ... Nomenclature While affected individuals have traditionally been classified as having one of three MPS I syndromes – Hurler syndrome, Hurler-Scheie syndrome, or Scheie syndrome – no biochemical differences have been identified and the clinical findings overlap; thus, affected individuals are best described as having either severe or attenuated MPS I, a distinction that influences therapeutic options.
There are three variants, differing widely in their severity, with Hurler syndrome being the most severe, Scheie syndrome the mildest and Hurler-Scheie syndrome giving an intermediate phenotype. Epidemiology Prevalence is estimated at 1/100,000, with Hurler syndrome accounting for 57% of cases, Hurler-Scheie syndrome accounting for 23% of cases and Scheie syndrome accounting for 20% of cases. ... Typical symptoms are stiff joints, corneal opacities, carpal tunnel syndrome and mild skeletal changes. Aortic valve disease can be present. ... The mutations result in complete deficiency of the enzyme in Hurler syndrome or partial function in Scheie syndrome, leading to lysosomal accumulation of dermatan sulfate (DS) and heparan sulfate (HS). ... Life expectancy is normal or only slightly affected in Scheie syndrome, but is reduced in Hurler syndrome, with death occurring before adolescence due to serious cardiovascular and respiratory complications.
Nomenclature McKusick et al. (1972) suggested that the Hurler syndrome might be called MPS IH and the Scheie syndrome MPS IS. ... The manifestations of Scheie syndrome are so mild that the diagnosis is often not considered until adulthood. ... The second case of Emerit et al. (1966) was probably Scheie syndrome. The parents were second cousins. ... The case of Poulet (1968) with 2 affected cousins was probably Scheie syndrome. Fischer et al. (1999) described combined aortic and mitral stenosis in a patient with the clinical diagnosis of Ullrich-Scheie syndrome. ... Population Genetics Lowry and Renwick (1971) reported the frequency of Scheie syndrome to be 1 in 500,000 births. Yamagishi et al. (1996) defined the IDUA mutations in 19 Japanese MPS I patients, including 2 pairs of sibs, with various clinical phenotypes; Hurler syndrome, 6 cases; Hurler/Scheie syndrome, 7 cases; Scheie syndrome, 6 cases.
Marfan syndrome most commonly affects the heart, eyes, blood vessels and skeleton. People with Marfan syndrome are usually tall and thin with unusually long arms, legs, fingers and toes. ... Most people with Marfan syndrome inherit the abnormal gene from a parent who has the disorder. ... Foot pain and low back pain are common with Marfan syndrome. Complications of pregnancy Marfan syndrome can weaken the walls of the aorta, the main artery that leaves the heart. ... In the past, people who had Marfan syndrome often died young. With regular monitoring and modern treatment, most people with Marfan syndrome can now expect to live a more normal life span.
The patient's father had Marfan syndrome with previous surgery for aortic dissection. The mother made the diagnosis of acute dissection in her daughter. The trisomy 8 syndrome (Pai et al., 1979) simulates the Marfan syndrome in its skeletal features. ... The diagnosis was clearly Marfan syndrome. Cistulli and Sullivan (1995) suggested that the Marfan syndrome is associated with a high prevalence of obstructive sleep apnea. ... Verbraecken et al. (1995) reported obstructive sleep hypopnea syndrome in a 35-year-old woman who had Marfan syndrome. ... Tekin et al. (2007) stated that this was the first report of familial neonatal Marfan syndrome. Voermans et al. (2009) evaluated 10 patients with Marfan syndrome specifically for neuromuscular features.
Marfan syndrome is a systemic disease of connective tissue characterized by a variable combination of cardiovascular, musculo-skeletal, ophthalmic and pulmonary manifestations. ... Etiology In the vast majority of cases, Marfan syndrome is caused by mutations of the FBN1 gene (15q21), which codes for fibrilline-1, a protein essential for connective tissues. ... Differential diagnosis Differential diagnoses include MASS syndrome, Shprintzen-Goldberg syndrome, mitral valve prolapse, Ehlers-Danlos syndrome and other diseases that present with aortic aneurysm such as Loeys-Dietz syndrome (see these terms).
Marfan syndrome is a disorder of the connective tissue. Connective tissue provides strength and flexibility to structures throughout the body such as bones, ligaments, muscles, walls of blood vessels, and heart valves. Marfan syndrome affects most organs and tissues, especially the skeleton, lungs, eyes, heart, and the large blood vessel that distributes blood from the heart to the rest of the body (the aorta). It is caused by mutations in the FBN1 gene, which provides instructions for making a protein called fibrillin-1. Marfan syndrome is inherited in an autosomal dominant pattern.
Nomenclature Outdated terms used in the description of Marfan syndrome include the following: Neonatal Marfan syndrome. ... Many of the skeletal features of Marfan syndrome are common in the general population. ... Up to 20% of individuals with TAAD who do not have features of Marfan syndrome, vascular Ehlers Danlos syndrome, or Loeys-Dietz syndrome have a family history of TAAD. Approximately 30% of families with HTAD who do not have a clinical diagnosis of Marfan syndrome or another syndrome have a causative pathogenic variant in one of the 13 known HTAD-related genes. ... The diagnosis of Stickler syndrome is clinically based. Stickler syndrome caused by a heterozygous pathogenic variant in COL2A1 , COL11A1 , or COL11A2 is inherited in an autosomal dominant manner; Stickler syndrome caused by biallelic pathogenic variants in COL9A1 , COL9A2 , or COL9A3 is inherited in an autosomal recessive manner.
Marfan syndrome is a disorder that affects the connective tissue in many parts of the body. ... Most individuals with Marfan syndrome have some degree of nearsightedness (myopia ). ... The features of Marfan syndrome can become apparent anytime between infancy and adulthood. ... Frequency The incidence of Marfan syndrome is approximately 1 in 5,000 worldwide. ... At least 25 percent of Marfan syndrome cases result from a new mutation in the FBN1 gene.
Lightwood-Albright syndrome Specialty Urology Lightwood–Albright syndrome is a neonatal form of renal tubular acidosis . [1] It is characterized by distal renal tubular acidosis that occurs as a result of bicarbonate wasting and the inability to excrete hydrogen ions. [2] [3] By definition, it is a transient process and has no particular disease course. If untreated, it may lead to nephrocalcinosis and failure to thrive . [4] It is also known as Lightwood Syndrome , [5] Butler-Albright Syndrome, [3] or Lightwood-Butler-Albright Syndrome. [6] It is named for Reginald Cyril Lightwood and Fuller Albright . [6] [7] [8] Contents 1 Demographic 2 Pathophysiology 3 Diagnosis 4 Treatment 5 References 6 External links Demographic [ edit ] Lightwood–Albright syndrome affects neonates . ... Specifically, the urine will be unable to reach a pH under 5.5 because of its basicity . [3] Clinical findings can include muscle wasting , vomiting, failure to thrive , fatigue, constipation, polyuria , and polydipsia . [4] The following are important differential diagnoses that should be considered by a medical provider before making the diagnosis. [4] Periodic Paralysis Adenosine-Deaminase Deficiency Gitelman Syndrome Lowe Syndrome Treatment [ edit ] Treatment of Lightwood–Albright syndrome is through transient alkali replacement therapy. [4] This treatment option utilizes alkali as a base to help equilibrate the amount of extra acid that is being retained in the body. ... PMID 13149190 . ^ a b c d e f g h Bissonnette, Bruno; Luginbuehl, Igor; Engelhardt, Thomas (2019), "Albright-Butler Syndrome" , Syndromes: Rapid Recognition and Perioperative Implications (2 ed.), New York, NY: McGraw-Hill Education , retrieved 2020-11-18 ^ a b c d e Bissonnette, Bruno; Luginbuehl, Igor; Engelhardt, Thomas (2019), "Lightwood Syndrome" , Syndromes: Rapid Recognition and Perioperative Implications (2 ed.), New York, NY: McGraw-Hill Education, ISBN 978-1-259-86178-9 , retrieved 2020-11-08 ^ " Lightwood syndrome " at Dorland's Medical Dictionary ^ a b c synd/993 at Who Named It? ... External links [ edit ] Classification D ICD - 10 : N25.8 v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy
Prune belly syndrome Other names Abdominal muscle deficiency syndrome, congenital absence of the abdominal muscles, Eagle-Barrett syndrome, [1] Obrinsky syndrome, [2] Fröhlich syndrome, [3] triad syndrome Prune belly syndrome in an Egyptian child with Down syndrome . ... Prune belly syndrome is a congenital disorder of the urinary system , characterized by a triad of symptoms. The syndrome is named for the mass of wrinkled skin that is often (but not always) present on the abdomen of those with the disorder. ... Another study that may suggest the syndrome is a voiding cystourethrogram . [ citation needed ] PBS is far more common in males. ... The orthopaedic manifestations of prune-belly (Eagle-Barrett) syndrome. J Bone Joint Surg Am. 77(2):251-7 ^ "Prune belly syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ^ synd/1499 at Who Named It?
A number sign (#) is used with this entry because of evidence that prune belly syndrome (PBS) is caused by homozygous mutation in the CHRM3 gene (118494) on chromosome 1q43. ... Futcher, knowing that I would be interested in it, sent for the child.') The full syndrome probably occurs only in males (Williams and Burkholder, 1967). A possibly related syndrome was described in a single patient by Texter and Murphy (1968). ... Nomenclature Dorst and Seidel (1982) suggested Eagle-Barrett syndrome as a 'kinder and more appropriate designation' than prune belly syndrome. ... There was no earlier family history of prune belly syndrome. Ramasamy et al. (2005) reviewed reported cases of 'complete' familial prune belly syndrome, in which the full clinical triad was present, noting that 28% of patients were female in the familial form of the syndrome compared to only 5% of nonfamilial cases.
Clinical description The spectrum ranges from mildly affected individuals to lethality. Prune belly syndrome (PBS) often presents antenatally on routine ultrasound with oligohydramnios and a very large, distended bladder, mild to severe bilateral hydroureteronephrosis, fetal ascites and, occasionally, renal dysplasia and a patent urachus. Other anomalies include cryptorchidism, pulmonary hypoplasia, club foot and features of Potter sequence. The syndrome is associated with pulmonary, skeletal, cardiac, and gastrointestinal defects and may be associated with atresia of the urethra. ... Differential diagnosis Differential diagnoses in utero include megacystis/megaureter, megacystis microcolon intestinal hypoperistalsis syndrome or posterior urethral valves. PBS has also been reported in trisomy 13, 18, 21 and large chromosome 6 deletions.
Prune belly syndrome (PBS) is a condition characterized by a lack of abdominal muscles, causing the skin on the abdominal area to wrinkle and appear "prune-like"; undescended testicles in males; and urinary tract malformations.
Exner officially named the disorder serpentine fibula polycystic kidney syndrome, but the term "Exner syndrome" became more prevalent. ... "SERPENTINE FIBULA-POLYCYSTIC KIDNEY SYNDROME" (PDF) . 600330 SERPENTINE FIBULA-POLYCYSTIC KIDNEY SYNDROME . ... "Serpentine fibula—polycystic kidney syndrome. A variant of the Melnick-Needles syndrome or a distinct entity?" ... "Further evidence that the Hajdu-Cheney syndrome and the "serpentine fibula-polycystic kidney syndrome" are a single entity" . ... "Serpentine fibula—polycystic kidney syndrome and Melnick-Needles syndrome are different disorders" .
Dejerine–Roussy syndrome Other names Thalamic pain syndrome Specialty Neurology Dejerine–Roussy syndrome or thalamic pain syndrome is a condition developed after a thalamic stroke , a stroke causing damage to the thalamus . [1] Ischemic strokes and hemorrhagic strokes can cause lesioning in the thalamus. [ citation needed ] As initial stroke symptoms (numbness and tingling) dissipate, an imbalance in sensation causes these later syndromes, characterizing Dejerine–Roussy syndrome. ... Although preliminary, these findings hold promise for an objective way to understand and treat patients with Dejerine–Roussy syndrome. [7] Diagnosis [ edit ] Dejerine-Roussy is a rare pain syndrome. ... The name Dejerine–Roussy syndrome was coined after their deaths. ... As of the early 2000s, longer treatments lasting months to years have been explored in the continued search for permanent removal of abnormal pain. [1] Eponym [ edit ] Dejerine–Roussy syndrome has also been referred to as: "Posterior Thalamic Syndrome", "Retrolenticular Syndrome", "Thalamic Hyperesthetic Anesthesia", "Thalamic Pain Syndrome", "Thalamic Syndrome", "Central Pain Syndrome", and "Central Post-Stroke Syndrome". [1] [17] [18] This condition is not associated with Roussy–Lévy syndrome or Dejerine–Sottas disease , both of which are genetic disorders . [19] [20] [21] See also [ edit ] Central post-stroke pain Central pain syndrome References [ edit ] ^ a b c d e f g h i j Klit, H., Finnerup, N. ... "Roussy–Lévy syndrome is a phenotypic variant of Charcot–Marie–Tooth syndrome IA associated with a duplication on chromosome 17p11.2".
Although antipsychotic drugs are known to be the main drugs that are concerned with this syndrome, several other drugs are reported to have caused the syndrome as well. ... In some cases, discontinuing the use of those drugs resulted in complete disappearance of the syndrome. The time it took for the improvement and the disappearance of the syndrome depended on the type of drug being administered or the specific cause of the syndrome itself. [10] Anticholinergic drugs [ edit ] Anticholinergic drugs have been reported to be extremely effective in 40% of the patients with the Pisa syndrome. [ citation needed ] Patients with Pisa syndrome that is resistant to anticholinergic drugs is mostly resolved by the reduction of the administration of the antipsychotic drugs as previously mentioned. ... Baba and H. Shiraishi, et al. The Pisa syndrome (pleurothotonus) during antidepressant therapy. ... Matsuzaka, et al. Drug-induced Pisa syndrome (pleurothotonus): epidemiology and management. ... Lindholm and L. Ljungerberg, New dystonic syndrome associated with butyrophenone therapy.
Lymphedema can also be a feature of syndromic disorders such as lymphedema-distichiasis syndrome (153400), which is caused by mutation in the FOXC2 gene (602402).
Milroy disease affects the lymphatic system and can lead to swelling ( lymphedema ) of the legs and feet. Prior to birth, symptoms of Milroy disease may include fluid buildup in the legs and feet and sometimes, within the body ( nonimmune hydrops ). After birth, symptoms may include swelling of the lower limbs, a buildup of fluid in the scrotum ( hydrocele ), and skin changes. People with Milroy disease have an increased risk for developing skin infections and certain types of cancer. Milroy disease occurs when the FLT4 gene is not working correctly. It is inherited in an autosomal dominant pattern.
A number sign (#) is used with this entry because of evidence that lymphatic malformation-4 (LMPHM4) is caused by heterozygous mutation in the VEGFC gene (601528) on chromosome 4q34. For a discussion of genetic heterogeneity of lymphatic malformation, see 153100. Clinical Features Gordon et al. (2013) reported a 3-generation Caucasian family in which 7 individuals had lymphedema affecting the lower limbs. The adult proband and his adult sister had congenital lymphedema of the feet and ankles, which in the sister alternately improved and deteriorated over time. Both had below-the-knee lymphedema; the sister also had prominent veins around the ankles and on the dorsa of the feet.
Milroy disease is a frequent form of primary lymphedema (see this term) characterized generally by painless, chronic lower-limb lymphedema found at birth or developing in the early neonatal period.
Milroy disease is a condition that affects the normal function of the lymphatic system. The lymphatic system produces and transports fluids and immune cells throughout the body. Impaired transport with accumulation of lymph fluid can cause swelling (lymphedema ). Individuals with Milroy disease typically have lymphedema in their lower legs and feet at birth or develop it in infancy. The lymphedema typically occurs on both sides of the body and may worsen over time.
Lymphedema in this syndrome affects the extremities and often improves over time. ... Noonan syndrome is characterized by short stature, congenital heart defect, and developmental delay of variable degree. ... Inheritance is autosomal dominant. People with Noonan syndrome may present with congenital lymphedema of the feet and legs. ... FOXC2 is the only gene known to be associated with lymphedema-distichiasis syndrome. Inheritance is autosomal dominant. ... Lymphedema with yellow nails (yellow nail syndrome, YNS) (OMIM 153300) often presents after age 50 years.
Description Hereditary primary lymphedema is caused by anatomic or functional defects in the lymphatic system, resulting in chronic swelling of body parts. There may be accompanying nail and skin changes, such as nail dysplasia or papillomatosis. Onset is usually at birth or in early childhood but can occur later, and the severity is variable (summary by Gordon et al., 2013 and Balboa-Beltran et al., 2014). For a discussion of genetic heterogeneity of lymphatic malformation, see 153100. Clinical Features Malik and Grzeschik (2008) reported a large consanguineous Pakistani family in which 25 individuals spanning 5 generations had lymphedema confined to the lower limbs.
A number sign (#) is used with this entry because of evidence that lymphatic malformation-3 (LMPHM3) is caused by heterozygous mutation in the GJC2 gene (608803) on chromosome 1q42. Description Hereditary primary lymphedema is caused by anatomic or functional defects in the lymphatic system, resulting in chronic swelling of body parts. There may be accompanying nail and skin changes, such as nail dysplasia or papillomatosis. Onset is usually at birth or in early childhood but can occur later, and the severity is variable (summary by Gordon et al., 2013 and Balboa-Beltran et al., 2014). For a discussion of genetic heterogeneity of lymphatic malformation, see 153100.
Primary lymphedema is a lymphatic system malformation characterized by swelling of an extremity that can be associated with other lymphatic effusions, due to an underlying developmental anomaly of the lymphatic system (abnormal lymphoangiogenesis). It can be hereditary or not and be congenital or late onset.
Some people with BAP1 tumor predisposition syndrome develop growths in the skin known as atypical Spitz tumors. People with this syndrome may have more than one of these tumors, and they can have dozens. ... However, individuals with malignant mesothelioma as part of the BAP1 tumor predisposition syndrome appear to survive longer than those who have the cancer without the syndrome. Frequency BAP1 tumor predisposition syndrome is a rare condition; its prevalence is unknown. ... Causes BAP1 tumor predisposition syndrome is caused by mutations in the BAP1 gene.
A number sign (#) is used with this entry because this tumor predisposition syndrome (TPDS) can be caused by heterozygous germline mutation in the BAP1 gene (603089) on chromosome 3p21. Description This tumor predisposition syndrome is inherited in an autosomal dominant pattern. ... Abdel-Rahman et al. (2011) concluded that this family had a hereditary cancer predisposition syndrome that increased the risk of several different types of tumors. The proband in this family was the only 1 of 53 unrelated patients with uveal melanoma and evidence of a familial cancer syndrome screened for BAP1 mutations who was found to carry a mutation, suggesting that is it a rare cause of hereditary uveal melanoma. ... In affected members of a large family with a tumor predisposition syndrome characterized mainly be renal cell carcinoma (RCC), Popova et al. (2013) identified a heterozygous germline mutation in the BAP1 gene (603089.0008).
BAP1-related tumor predisposition syndrome (TPDS) is an inherited cancer-predisposing syndrome, associated with germline mutations in BAP1 tumor suppressor gene.
For the anatomical abnormality observed in 1965, see Fraser syndrome . Frasier syndrome Specialty Endocrinology , obstetrics and gynaecology , urology , medical genetics Frasier syndrome is a urogenital anomaly associated with the WT1 (Wilms tumor 1 gene) gene. [1] [2] [3] It was first characterized in 1964. [4] Contents 1 Presentation 2 Genetics 2.1 Inheritance pattern 3 Diagnosis 4 Treatment 5 References 6 External links Presentation [ edit ] Frasier syndrome presents at birth with male pseudohermaphroditism (the external genitalia have a female appearance despite an XY genotype ), streak gonads and progressive glomerulonephropathy (focal segmental glomerulosclerosis ). ... The glomerulonephropathy presents later than in Denys-Drash syndrome , and the tumour risk phenotype is different; whilst Denys-Drash syndrome is associated with Wilms' tumour , Frasier syndrome is associated with gonadoblastoma . ... PMID 9499425 . ^ Reference, Genetics Home. "Frasier syndrome" . Genetics Home Reference . Retrieved 2018-04-17 . ^ "Frasier syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ... "Donor splice-site mutations in WT1 are responsible for Frasier syndrome". Nature Genetics . 17 (4): 467–70. doi : 10.1038/ng1297-467 . PMID 9398852 . ^ a b "Frasier syndrome" . Genetics Home Reference . U.S.
Frasier syndrome is a condition that affects the kidneys and genitalia. Frasier syndrome is characterized by kidney disease that begins in early childhood. ... Because they do not have all the features of the condition, females are usually given the diagnosis of isolated nephrotic syndrome. Frequency Frasier syndrome is thought to be a rare condition; approximately 50 cases have been described in the scientific literature. ... The WT1 gene mutations that cause Frasier syndrome lead to the production of a protein with an impaired ability to control gene activity and regulate the development of the kidneys and reproductive organs, resulting in the signs and symptoms of Frasier syndrome. Frasier syndrome has features similar to another condition called Denys-Drash syndrome, which is also caused by mutations in the WT1 gene.
Clinical Features Moorthy et al. (1987) suggested that some of the patients reported as cases of Denys-Drash syndrome (194080) in fact had a different disorder for which they suggested the designation Frasier syndrome (Frasier et al., 1964). ... Melo et al. (2002) reported a patient diagnosed with Frasier syndrome who had the external genitalia characteristic of Denys-Drash syndrome. ... Barbaux et al. (1997) suggested that the patient reported as having Denys-Drash syndrome and a mutation in intron 5 of the WT1 gene (607102.0009) in fact had Frasier syndrome. ... In the Denys-Drash syndrome, the tumor risk is much greater than in Frasier syndrome. ... The authors concluded that this patient had the external genitalia characteristic of Denys-Drash syndrome, suggesting that these 2 syndromes are not distinct diseases but may represent 2 ends of a spectrum of disorders caused by alterations in the WT1 gene.
A rare genetic, syndromic glomerular disorder characterized by the association of progressive glomerular nephropathy and 46,XY complete gonadal dysgenesis with a high risk of developing gonadoblastoma. ... It develops during childhood presenting as persistent proteinuria and subsequently steroid-resistant nephrotic syndrome (SRNS) and progresses to end-stage renal disease (ESRD) in the second or third decade of life. ... Wilms tumor is not common in individuals with Frasier syndrome. Etiology Frasier syndrome has been associated to specific pathogenic variants affecting nucleotides 4-5 of the intron 9 (previously referred to as IVS9+4; IVS9+5) in the WT1 gene (11p13). ... Differential diagnosis The main differential diagnosis is idiopathic steroid-resistant nephrotic syndrome, and other WT1 associated diseases including Denys-Drash syndrome, genetic steroid resistant nephrotic syndrome and disorders of testicular development. ... After kidney transplantation, nephrotic syndrome does not recur. 46,XY individuals with complete gonadal dysgenesis are infertile. * European Reference Network
Mutations in the same gene cause Gilbert syndrome (143500) and Crigler-Najjar syndrome type I (218800). Description The hereditary hyperbilirubinemias (Wolkoff et al., 1983) include (1) those resulting in predominantly unconjugated hyperbilirubinemia: Gilbert or Arias syndrome, Crigler-Najjar syndrome type I, and Crigler-Najjar syndrome type II; and (2) those resulting in predominantly conjugated hyperbilirubinemia: Dubin-Johnson syndrome (237500), Rotor syndrome (237450), and several forms of intrahepatic cholestasis (147480, 211600, 214950, 243300). ... Inheritance The inheritance pattern of Crigler-Najjar syndrome type II is generally considered to be autosomal recessive (Chowdhury et al., 2001). ... Guldutuna et al. (1995) described Crigler-Najjar syndrome type II in a 34-year-old Turkish woman, the daughter of first-cousin parents. ... Both of these variants had previously been found in patients with Gilbert syndrome. The authors speculated that the phenotype of Crigler-Najjar syndrome type II in this patient was caused by a combination of the heterozygous missense and homozygous insertion mutations.
A hereditary disorder of bilirubin metabolism characterized by unconjugated hyperbilirubinemia due to reduced and inducible activity of hepatic bilirubin glucuronosyltransferase (GT). Crigler-Najjar syndrome type 2 (CNS2) is a milder form of Crigler-Najjar syndrome (CNS) than Crigler-Najjar syndrome type 1 (CNS1). ... Differential diagnosis Differential diagnosis includes disorders of excessive bilirubin production (hemolysis) and impaired hepatic handling of bilirubin (hepatitis and Gilbert syndrome; see this term). CNS2 can be differentiated from CNS1 by measurement of transferase activity and the response to phenobarbital treatment.
Crigler-Najjar syndrome type 2 (CN-2) is a rare disorder that causes elevated levels of bilirubin in the blood (hyperbilirubinemia). ... Of note, mutations in the UGT1A1 gene can alternatively cause other disorders, such as Crigler-Najjar syndrome type 1 (CN-1) and Gilbert syndrome . ... Phenobarbitol treatment is ineffective for people with CN-1, which is treated differently. Gilbert syndrome is considered a mild liver disorder that often does not cause symptoms or causes mild jaundice. Sometimes it can be hard to distinguish between Gilbert syndrome and CN-2 because of considerable overlap in measured bilirubin levels.
The formation of a blood clot within the hepatic veins can lead to Budd–Chiari syndrome. The syndrome can be fulminant , acute, chronic, or asymptomatic. ... The fulminant syndrome presents early with encephalopathy and ascites. ... Liver biopsy is nonspecific but sometimes necessary to differentiate between Budd–Chiari syndrome and other causes of hepatomegaly and ascites, such as galactosemia or Reye's syndrome . ... Archived from the original on 2013-01-05. ^ "Etiology, management, and outcome of the Budd–Chiari syndrome". Cite journal requires |journal= ( help ) ^ "Hepatic vein thrombosis (Budd–Chiari syndrome)". Cite journal requires |journal= ( help ) ^ "The Budd–Chiari syndrome: a review". Cite journal requires |journal= ( help ) ^ "Budd–Chiari syndrome: long-term survival and factors affecting mortality".
Budd-Chiari syndrome is a rare disorder characterized by obstruction of the veins of the liver that carry the blood flow from the liver. ... In about one third of the cases, the cause of Budd-Chiari syndrome is unknown. Drugs or surgical interventions may be used to dissolve or decrease the size of the obstruction (if it is a clot). In some cases liver transplantation is needed. Budd-Chiari syndrome should be considered separate from veno-occlusive disease (VOD).
One of the causes of the Budd-Chiari syndrome is a membranous obstruction of the inferior vena cava (MOVC). ... Molecular Genetics Mahmoud et al. (1997) reported the incidence of the factor V Leiden mutation (R506Q; 612309.0001) in Budd-Chiari syndrome and portal vein thrombosis. The R506Q mutation was seen in 7 (23%) of 30 patients with Budd-Chiari syndrome (6 heterozygotes and 1 homozygote), 3 of whom had coexistent myeloproliferative disease. ... Gurakan et al. (1999) described a child with Budd-Chiari syndrome who was homozygous for the factor V Leiden mutation. Budd-Chiari syndrome is rare in children. Janssen et al. (2000) compared 43 patients with Budd-Chiari syndrome and 92 patients with portal vein thrombosis with 474 population-based controls. ... The relative risk of Budd-Chiari syndrome or portal vein thrombosis was not increased in the presence of inherited protein S or antithrombin deficiency.
Congenital condition of brain, cardiovascular and eye abnormalities PHACE Syndrome Other names Pascual-Castroviejo type II syndrome, P-CIIS, Pascual-Castroviejo syndrome type 2 [1] Specialty Medical genetics PHACE syndrome is a cutaneous condition characterized by multiple congenital abnormalities. [2] [3] The mnemonic PHACE stands for P osterior fossa brain malformations, H emangioma , A rterial lesions, C ardiac abnormalities, and E ye abnormalities. [4] PHACE syndrome should be considered in infants with large plaque-type facial hemangiomas. [5] Children presenting with this dermatologic manifestation should receive careful ophthalmologic, cardiac, and neurologic assessment. ... The team of experts pay close attention to how these children develop throughout the school age period. [11] [12] PHACE syndrome Handbook - Dr. Beth Drolet [13] In 2013, the PHACE syndrome Community was formed. ... PMID 19101041 . ^ "PHACE Syndrome" . Cincinnati Children's Hospital Medical Center . ... Retrieved 2018-10-25 . ^ "PHACE syndrome" . Children's Hospital of Wisconsin . ... Retrieved 2018-10-25 . ^ "PHACE Syndrome Community" . ^ Drolet, Beth. "PHACE Syndrome" .