M. (2003). "Tissue-specific Cushing's syndrome, 11beta-hydroxysteroid dehydrogenases and the redefinition of corticosteroid hormone action" (PDF) . ... J. (2012). "11β-Hydroxysteroid dehydrogenase type 1: Relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus". ... "Mutations of the hexose-6-phosphate dehydrogenase gene rarely cause hyperandrogenemic polycystic ovary syndrome" . Steroids . 76 (1–2): 135–9. doi : 10.1016/j.steroids.2010.10.001 . ... M. (2003). "Tissue-specific Cushing's syndrome, 11beta-hydroxysteroid dehydrogenases and the redefinition of corticosteroid hormone action" (PDF) . ... "The administration of estrogens, combined with anti-androgens, has beneficial effects on the hormonal features and asymmetric dimethyl-arginine levels, in women with the polycystic ovary syndrome". Atherosclerosis . 196 (2): 958–65. doi : 10.1016/j.atherosclerosis.2007.03.002 .
A number sign (#) is used with this entry because of evidence that cortisone reductase deficiency-2 (CORTRD2) is caused by heterozygous mutation in the HSD11B1 gene (600713) on chromosome 1q32. Description Cortisone reductase deficiency (CRD) is a disorder in which there is a failure to regenerate the active glucocorticoid cortisol from cortisone via the enzyme 11-beta-hydroxysteroid dehydrogenase, encoded by the HSD11B1 gene. Purified 11-beta-HSD acts readily as a dehydrogenase, inactivating cortisol to cortisone; however, in the presence of a high NADPH/NADP+ ratio, generated in vivo through the activity of microsomal hexose-6-phosphate dehydrogenase (H6PD; 138090), 11-beta-HSD switches to ketoreductase activity and generates active glucocorticoid. Lack of cortisol regeneration stimulates ACTH-mediated adrenal hyperandrogenism, with males manifesting in early life with precocious pseudopuberty and females presenting later with hirsutism, oligomenorrhea, and infertility. Biochemically, CRD is diagnosed through the assessment of urinary cortisol and cortisone metabolites and consists of measuring the ratio of tetrahydrocortisol (THF) plus 5-alpha-THF to tetrahydrocortisone (THE), which in CRD patients is typically less than 0.1 (reference range, 0.7 to 1.2) (summary by Lawson et al., 2011).
Genetic Heterogeneity of Cortisone Reductase Deficiency CORTRD2 (614662) is caused by mutation in the HSD11B1 gene (600713) on chromosome 1q32. Clinical Features A syndrome consistent with type I 11-beta-hydroxysteroid dehydrogenase (HSD11B1) deficiency had been described in 3 female patients, 2 of whom were sibs (Taylor et al., 1984; Phillipou and Higgins, 1985; Savage et al., 1991; Phillipov et al., 1996). ... In the absence of a confirmed defect in the HSD11B1 gene, Phillipov et al. (1996) termed the syndrome 'apparent cortisone reductase deficiency.'
A rare, genetic, endocrine disease characterized by defect in conversion of cortisone to active cortisol, resulting in ACTH-mediated excessive androgen release from adrenal glands. Premature adrenarche is typical with precocious pseudopuberty, proportionate tall stature and accelerated bone maturation in males, and hirsutism, oligoamenorrhea, central obesity and infertility in females. Imaging studies may indicate adrenal hyperplasia.
These studies suggest that the dis-regulated overproduction of interleukin 5 by blood mononuclear cells is an underlying cause of the eosinophilia found in at least some families with this disorder. [6] Diagnosis [ edit ] The diagnosis of familial eosinophilia rest upon a) familial clustering of the disorder; b) exclusion of "family acquired eosinophilia" (i.e. eosinophilia due to chronic parasite or other infestations that afflict multiple members of a family); c) lack of eosinophil-induced tissue destruction such as that which occurs in the hypereosinophilic syndrome ; d) absence of signs or symptoms of other hereditary eosinophilias (e.g. hyperimmunoglobulin E syndrome , Omenn syndrome ), Wiskott–Aldrich syndrome , and numerous other primary immunodeficiency syndromes); and d) lack of evidence for reactive eosinophilias (e.g. allergy -related), neoplastic eosinophilias (e.g. eosinophilic leukemia , clonal eosinophilia ), or eosinophila associated with various hematological and non-hematological malignancies as outlined in causes of eosinophilia . [2] [3] [5] [10] [11] Treatment [ edit ] Since longitudinal data, including eosinophil counts obtained over a 20- to 30-year periods in some family members, show remarkable stability in the absolute eosinophil count with no evidence of disease attributable to eosinophil-related tissue injury, therapy to lower eosinophil counts such as corticosteroids , tyrosine kinase inhibitors , and antibodies directed against interleukin 5 have in general not been used to treat familial eosinophilia. ... "Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes" . The Journal of Allergy and Clinical Immunology . 130 (3): 607–612.e9. doi : 10.1016/j.jaci.2012.02.019 . ... PMID 26209891 . ^ Curtis C, Ogbogu P (2016). "Hypereosinophilic Syndrome". Clinical Reviews in Allergy & Immunology . 50 (2): 240–51. doi : 10.1007/s12016-015-8506-7 .
Cortical blindness Specialty Neurology Cortical blindness is the total or partial loss of vision in a normal-appearing eye caused by damage to the brain 's occipital cortex . [1] Cortical blindness can be acquired or congenital, and may also be transient in certain instances. [2] Acquired cortical blindness is most often caused by loss of blood flow to the occipital cortex from either unilateral or bilateral posterior cerebral artery blockage ( ischemic stroke ) and by cardiac surgery. [2] In most cases, the complete loss of vision is not permanent and the patient may recover some of their vision ( cortical visual impairment ). [2] Congenital cortical blindness is most often caused by perinatal ischemic stroke, encephalitis , and meningitis . [3] Rarely, a patient with acquired cortical blindness may have little or no insight that they have lost vision, a phenomenon known as Anton–Babinski syndrome . Cortical blindness and cortical visual impairment (CVI), which refers to the partial loss of vision caused by cortical damage, are both classified as subsets of neurological visual impairment (NVI). ... One diagnostic marker of this distinction is that the pupils of individuals with cortical blindness will respond to light whereas those of individuals with ocular visual impairment will not. [ citation needed ] Contents 1 Symptoms 2 Causes 3 Diagnosis 4 Outcome 5 See also 6 References 7 Further reading 7.1 Books 7.2 Papers 8 External links Symptoms [ edit ] The most common symptoms of acquired and transient cortical blindness include: A complete loss of visual sensation and of vision [4] Preservation/sparing of the abilities to perceive light and/or moving, but not static objects ( Riddoch syndrome ) [2] A lack of visual fixation and tracking [4] Denial of visual loss ( Anton–Babinski syndrome ) Visual hallucinations [4] Macular sparing, in which vision in the fovea is spared from the blindness. [4] Causes [ edit ] The most common cause of cortical blindness is ischemia ( oxygen deprivation ) to the occipital lobes caused by blockage to one or both of the posterior cerebral arteries. [2] However, other conditions have also been known to cause acquired and transient cortical blindness, including: Congenital abnormalities of the occipital lobe [5] Head trauma to the occipital lobe of the brain [5] Bilateral lesions of the primary visual cortex [2] Infection [5] Creutzfeldt–Jakob disease (CJD), in association with a rapid onset of dementia rarely Dissociative identity disorder (DID) [6] Side effect of some anti-epilepsy drugs ( AEDs ) Hyperammonemia [7] Eclampsia [8] and, rarely, pre-eclampsia [9] [10] The most common causes of congenital cortical blindness are: Traumatic brain injury (TBI) to the occipital lobe of the brain [3] Congenital abnormalities of the occipital lobe [5] Perinatal ischemia [3] Encephalitis [3] Meningitis [3] Diagnosis [ edit ] A patient with cortical blindness has no vision but the response of his/her pupil to light is intact (as the reflex does not involve the cortex). ... Cortical blindness can be associated with visual hallucinations , denial of visual loss ( Anton–Babinski syndrome ), and the ability to perceive moving but not static objects. ( Riddoch syndrome ). [ citation needed ] Outcome [ edit ] The prognosis of a patient with acquired cortical blindness depends largely on the original cause of the blindness. ... External links [ edit ] Classification D ICD - 10 : H47.6 ICD - 10-CM : H47.61 ICD - 9-CM : 377.75 MeSH : D019575 SNOMED CT : 68574006 v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis
If the nerves lost to denervation are part of the neuronal communication to a specific function in the body then altered or a loss of physiological functioning can occur. [1] Denervation can be caused by injury or be a symptom of a disorder like ALS and post-polio syndrome . Additionally, it can be a useful surgical technique to alleviate major negative symptoms, such as in renal denervation . ... This plasticity allows for the brain to compensate for the loss in neuronal communication resulting from injury. [3] Disorders [ edit ] Denervation processes have a strong association with the symptoms seen in post-polio syndrome . Those with post polio syndrome are undergoing a constant process of denervation and reinnervation . ... Following an acute polio infection diagnosis symptoms such as fatiguability , general weakness and pain are believed to be correlated to muscle denervation. [4] Much like post-polio syndrome, amyotrophic lateral sclerosis also has similar symptoms of motor neuron degeneration leading to general weakness and in some cases paralysis. ... Entrapment and compressive neuropathy syndromes occur due to compression and/or constriction on a specific location for a segment of a single nerve or multiple nerve sites. ... ISSN 1098-2779 . ^ a b Gonzalez, Henrik; Olsson, Tomas; Borg, Kristian (June 2010). "Management of postpolio syndrome". The Lancet Neurology . 9 (6): 634–642. doi : 10.1016/s1474-4422(10)70095-8 .
Some evidence has indicated TH2 lymphocytes may also play a role, but further investigation is needed. [ citation needed ] Kimura's disease is generally limited to the skin, lymph nodes, and salivary glands, but patients with Kimura's disease and nephrotic syndrome have been reported. The basis of this possible association is unclear. [1] Diagnosis [ edit ] An open biopsy is the chief means by which this disease is diagnosed. [ citation needed ] "Lymphoid nodules with discrete germinal centers can occupy an area extending from the reticular dermis to the fascia and muscle. ... External links [ edit ] Classification D ICD - 10 : I89.8 MeSH : D000796 DiseasesDB : 31710 External resources eMedicine : article/1098777 Orphanet : 482 v t e Neutrophilic and eosinophilic dermatoses Eosinophilic dermatosis With vasculitis Eosinophilic vasculitis Eosinophilic granulomatosis with polyangiitis Without vasculitis Arthropod assault Eosinophilic cellulitis Hypereosinophilic syndrome Papuloerythroderma of Ofuji Granuloma faciale Eosinophilic folliculitis Ungrouped Angiolymphoid hyperplasia with eosinophilia / Kimura's disease Annular erythema of infancy Eosinophilic fasciitis Eosinophilic granuloma Eosinophilic ulcer of the oral mucosa Erythema toxicum neonatorum Incontinentia pigmenti Itchy red bump disease Juvenile xanthogranuloma Pachydermatous eosinophilic dermatitis Papular eruption of blacks Pruritic papular eruption of HIV disease Reactive neutrophilic dermatoses Epidermis Keratoderma blennorrhagicum Subcorneal pustular dermatosis Dermis without vasculitis : Sweet's syndrome Pyoderma gangrenosum Bowel-associated dermatosis–arthritis syndrome with vasculitis: Neutrophilic dermatosis of the dorsal hands Ungrouped Acute erythema nodosum Marshall syndrome Neutrophilic eccrine hidradenitis Pyogenic arthritis–pyoderma gangrenosum–acne syndrome Rheumatoid neutrophilic dermatitis Superficial granulomatous pyoderma Sweet's syndrome-like dermatosis Vesicopustular dermatosis
Kimura disease is a benign and chronic inflammatory disorder of unknown etiology, occurring mainly in Asian countries (very rarely in Western countries) and predominantly affecting young men, that usually presents with solitary or multiple non-tender subcutaneous masses in the head and neck region (in particular the preauricular and submandibular area) and/or generalized painless lymphadenopathy, often with salivary gland involvement. Characteristic laboratory findings include blood eosinophilia and markedly elevated serum immunoglobulin E (IgE) levels. It is often associated with autoinflammatory disorders (i.e. ulcerative colitis, bronchial asthma) and a co-existing renal disease.
Kimura disease is a rare, noncancerous, chronic condition that causes the tissue under the skin of the head or neck to become swollen. Masses generally appear in a person's mid-20s and the disease mainly affects Asian men. Kimura disease is diagnosed by a surgical biopsy and the cause of the disease is unknown. Treatment includes surgery, medication, or a combination of the two. However, the masses tend to develop again after treatment. Because this condition rarely causes health problems, people with Kimura disease have a good outcome.
A number sign (#) is used with this entry because of evidence that congenital anomalies of the kidney and urinary tract syndrome with or without hearing loss, abnormal ears, or developmental delay (CAKUTHED) is caused by heterozygous mutation in or deletion of the PBX1 gene (176310) on chromosome 1q23. Some individuals with the CAKUTHED phenotype have deletion of several genes in this region, consistent with a contiguous gene deletion syndrome. Description CAKUTHED is an autosomal dominant highly pleiotropic developmental disorder characterized mainly by variable congenital anomalies of the kidney and urinary tract, sometimes resulting in renal dysfunction or failure, dysmorphic facial features, and abnormalities of the outer ear, often with hearing loss. ... Clinical Features Le Tanno et al. (2017) reported 8 unrelated patients with a syndromic form of CAKUT associated with variable deletions of chromosome 1q23.3-q24.1; the patients were collected through several microarray databases with a focus on CAKUT. ... INHERITANCE - Autosomal dominant GROWTH Other - Growth retardation HEAD & NECK Face - Dysmorphic facial features, variable - Long face - Narrow face - Prominent philtrum Ears - Low-set ears - Abnormally shaped ears - Hypoplastic ears - Anteverted ears - Crumpled ears - Abnormal ear lobes - Thickened helices - Hypoplastic helices - Hearing loss (in some patients) Eyes - Epicanthal folds - Strabismus Nose - Broad nasal bridge - Anteverted nares Mouth - Thin upper lip CARDIOVASCULAR Heart - Congenital heart defects (in some patients) RESPIRATORY - Respiratory insufficiency (in some patients) CHEST Diaphragm - Diaphragmatic hernia ABDOMEN Gastrointestinal - Poor feeding GENITOURINARY - Ambiguous genitalia - Abnormal sexual development External Genitalia (Male) - Cryptorchidism - Micropenis Kidneys - Renal hypoplasia - Renal dysplasia - Renal ectopia - Horseshoe kidney - Renal agenesis - Renal insufficiency - Renal pelvis dilatation - Hyperechogenic kidneys - Cystic dysplasia - Poor corticomedullary demarcation - Oligonephronia - Renal failure (in some patients) Ureters - Urinary tract abnormalities - Bifid ureter - Absent ureter Bladder - Vesicoureteral reflux SKIN, NAILS, & HAIR Skin - Sacral pit (in some patients) MUSCLE, SOFT TISSUES - Hypotonia NEUROLOGIC Central Nervous System - Developmental delay (in some patients) - Motor delay - Speech delay PRENATAL MANIFESTATIONS Amniotic Fluid - Oligohydramnios MISCELLANEOUS - Onset at birth or early infancy - Highly variable phenotype - Extra-renal manifestations are variable - Variable severity - De novo mutation - Some patients have a contiguous gene deletion syndrome involving the PBX1 gene MOLECULAR BASIS - Caused by mutation in the pre-B-cell leukemia transcription factor 1 gene (PBX1, 176310.0001 ) ▲ Close
Basal cell carcinoma occurs as a feature of multiple syndromes, including basal cell nevus syndrome (BCNS; 109400), Bazex syndrome (301845), Rombo syndrome (180730), Brooke-Spiegler syndrome (605041), Muir-Torre syndrome (158320), and xeroderma pigmentosum (see 278700). ... Inheritance Happle (2000) postulated that there is an autosomal dominant phenotype characterized by multiple superficial BCC without associated anomalies that is distinct from nevoid basal cell carcinoma syndrome (109400). The author cited 2 lines of evidence in favor of this hypothesis.
The PRODH gene falls within the region deleted in the 22q11 deletion syndrome, including DiGeorge syndrome (188400) and velocardiofacial syndrome (192430). ... The patient exhibited symptoms of DiGeorge syndrome, a contiguous gene deletion syndrome involving 22q11, and also appeared to be heterozygous for deletion of the heparin cofactor II gene (HCF2; 142360), which maps to 22q11. ... Goodman et al. (2000) reported that 8 of 15 patients with deletions in the velocardiofacial syndrome critical region 22q11.2 had elevated proline levels ranging from 278 to 849 micromol/l while 7 were in the normal range of 51 to 271 micromol/l. ... Goodman et al. (2000) concluded that elevations in plasma proline levels should be considered a biochemical feature of the chromosome 22q11.2 deletion syndromes and suggested that patients with isolated hyperprolinemia should be studied for the microdeletion at 22q11.2.
A rare disorder of proline metabolism characterized biochemically by markedly elevated levels of proline in plasma and urine due to deficiency of proline oxidase. The reported clinical phenotype ranges from asymptomatic to variable neurologic and psychiatric manifestations (including global developmental delay, seizures, autistic features, and hyperactivity).
Hyperprolinemia is an excess of a particular protein building block (amino acid), called proline, in the blood. This condition generally occurs when proline is not broken down properly by the body. There are two inherited forms of hyperprolinemia, called type I and type II. People with hyperprolinemia type I often do not show any symptoms, although they have proline levels in their blood between 3 and 10 times the normal level. Some individuals with hyperprolinemia type I exhibit seizures, intellectual disability, or other neurological or psychiatric problems.
Hyperprolinemia is when there is an excess of a particular protein building block (amino acid), called proline, in the blood. This condition generally occurs when proline is not broken down properly by the body. There are two inherited forms: hyperprolinemia type 1 and hyperprolinemia type 2 . People with hyperprolinemia type I often do not show any symptoms, although they have proline levels in their blood between 3 and 10 times the normal level. Less commonly, affected individuals can experience seizures, intellectual disability, or other neurological or psychiatric problems.
Congenital protein C deficiency is an inherited coagulation disorder characterized by deep venous thrombosis symptoms due to reduced synthesis and/or activity levels of protein C. Epidemiology Prevalence of severe protein C deficiency (homozygous or compound heterozygous forms) is estimated at 1/ 500,000. Partial deficiencies (heterozygous forms) are much more frequent (1/200-1/500). Men and women are equally affected. Clinical description Patients with undetectable protein C levels usually manifest the disease several hours to days after birth, with purpura fulminans (see this term) or massive venous thrombosis. Purpura fulminans is a life-threatening condition involving severe clotting throughout the body and causing necrosis of tissues.
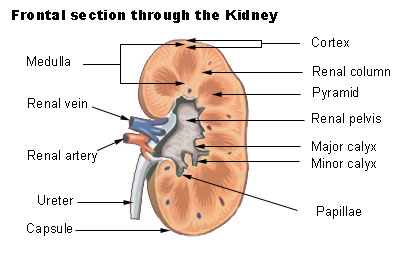
This leads to the clinical features of dRTA: [1] Normal anion gap metabolic acidosis /acidemia Hypokalemia Urinary stone formation (related to alkaline urine, hypercalciuria , and low urinary citrate). [2] Nephrocalcinosis (deposition of calcium in the substance of the kidney) Bone demineralisation (causing rickets in children and osteomalacia in adults) The symptoms and sequelae of dRTA are variable and range from being completely asymptomatic , to loin pain and hematuria from kidney stones , to failure to thrive and severe rickets in childhood forms as well as possible renal failure and even death. dRTA commonly leads to sodium loss and volume contraction, which causes a compensatory increase in blood levels of aldosterone . [3] Aldosterone causes increased resorption of sodium and loss of potassium in the collecting duct of the kidney, so these increased aldosterone levels cause the hypokalemia which is a common symptom of dRTA. [3] Causes [ edit ] Diagram depicting an alpha intercalated cell with the apical proton pump and basolateral band 3 (kAE1) Autoimmune disease . Classically Sjögren's syndrome , but it is also associated with systemic lupus erythematosus , rheumatoid arthritis and even hypergammaglobulinemia . ... External links [ edit ] Classification D OMIM : 179800 MeSH : D000141 External resources MedlinePlus : 000493 Orphanet : 18 v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy v t e Acid–base disorders Acidosis Metabolic High anion gap Ketoacidosis Diabetic ketoacidosis Alcoholic ketoacidosis Lactic Normal anion gap Hyperchloremic Renal tubular Respiratory Respiratory Alkalosis Metabolic Contraction alkalosis Respiratory Other Mixed disorder of acid-base balance Acid–base homeostasis
Acquired forms of dRTA are thought to be caused by autoimmune diseases such as Sjögren syndrome (see this term) or secondary to other conditions like sickle cell anemia, systemic lupus erythematosus (see these terms), chronic obstructive uropathy, or post-renal transplantation.
A rare inherited form of distal renal tubular acidosis (dRTA) characterized by hyperchloremic metabolic acidosis often but not always associated with hypokalemia. Epidemiology The prevalence is unknown. Clinical description Disease onset occurs in adolescence or adulthood and initial manifestations can include polyuria, polydipsia, muscle weakness and fatigue. Osteomalacia or osteopenia can occur due to calcium salt loss from the bones. Hypercalciuria, nephrolithiasis and nephrocalcinosis may result from long term chronic metabolic acidosis. Renal failure has not been described. Etiology AD dRTA is due to mutations in the SLC4A1 gene (17q21.31) encoding the band 3 anion transport protein (AE1).
SLC4A1 -associated distal renal tubular acidosis is a kidney (renal) disorder that sometimes includes blood cell abnormalities. The kidneys normally filter fluid and waste products from the body and remove them in urine; however, in people with distal renal tubular acidosis, the kidneys are unable to remove enough acid from the body, and the blood becomes too acidic. This chemical imbalance is called metabolic acidosis. The inability to remove acids from the body often results in slowed growth and may also lead to softening and weakening of the bones, called rickets in children and osteomalacia in adults. This bone disorder is characterized by bone pain, bowed legs, and difficulty walking. In addition, most children and adults with SLC4A1 -associated distal renal tubular acidosis have excess calcium in the urine (hypercalciuria), calcium deposits in the kidneys (nephrocalcinosis), and kidney stones (nephrolithiasis).
A number sign (#) is used with this entry because autosomal dominant distal renal tubular acidosis is caused by heterozygous mutation in the SLC4A1 gene (109270) on chromosome 17q21. Clinical Features Randall and Targgart (1961) observed renal tubular acidosis in members of several successive generations. All affected members showed both acidosis and nephrocalcinosis. Randall (1967) provided follow-up of this family. The pedigree included 4 instances of male-to-male transmission. The features were nephrocalcinosis, fixed urinary specific gravity, fixed urinary pH of about 5.0, high serum chloride, low serum bicarbonate, osteomalacia, and hypocalcemia. Alkalinization was effective therapy. Seedat (1968) observed 18 affected persons in 3 generations.
External links [ edit ] Classification D ICD - 10 : N17.2 ICD - 9-CM : 584.7 MeSH : D007681 DiseasesDB : 9572 External resources MedlinePlus : 000488 eMedicine : med/2839 radio/523 Scholia has a topic profile for Renal papillary necrosis . v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
Visual impairment Visual snow syndrome Other names Persistent positive visual phenomenon, [1] visual static, aeropsia Animated example of visual snow-like noise Specialty Neurology , Neuro-ophthalmology Symptoms Visual Snow , Palinopsia , Blue field entoptic phenomenon , Photophobia , Nyctalopia Usual onset Early to middle adulthood [1] Causes Unknown [2] Differential diagnosis Migraine aura , [3] Persistent aura without infarction Visual snow , also known as visual static , is a condition in which people see white or black dots in parts or the whole of their visual fields . [3] [4] The condition is typically always present and can last years. [5] The cause of visual snow is unclear. [2] Those affected typically also have migraines . [3] [4] The underlying mechanism is believed to involve excessive excitability of neurons in the right lingual gyrus and left cerebellar anterior lobe of the brain . [4] [6] Research has been limited because of issues of case identification and diagnosis, the latter now largely addressed, and the limited size of any studied cohort. ... However, comorbid migraine worsens some of the additional visual symptoms and tinnitus seen in "visual snow" syndrome. This might bias research studies by patients with migraine being more likely to offer study participation than those without migraine due to having more severe symptoms. ... Hallucinogen persisting perception disorder (HPPD), a condition caused by hallucinogenic drug use, is sometimes linked to visual snow, [12] but both the connection of visual snow to HPPD [13] and the cause and prevalence of HPPD is disputed. [14] Most of the evidence for both is generally anecdotal, and subject to spotlight fallacy. [13] [14] Diagnosis [ edit ] Proposed diagnostic criteria for the "visual snow" syndrome: [15] [16] Dynamic, continuous, tiny dots in the entire visual field. ... "Visual snow: a potential cortical hyperexcitability syndrome". Current Treatment Options in Neurology . 19 (3): 9. doi : 10.1007/s11940-017-0448-3 . ... External links [ edit ] Visual snow syndrome at NIH http://visualsnowsyndrome.com/ Classification D ICD - 10 : H53.1 v t e Phenomena of the visual system Entoptic phenomena Blind spot Phosphene Floater Afterimage Haidinger's brush Prisoner's cinema Blue field entoptic phenomenon Purkinje images Other phenomena Aura Form constant Scintillating scotoma Palinopsia Visual snow Afterimage on empty shape Cosmic ray visual phenomena Scotopic sensitivity syndrome Closed-eye hallucination
Morton's toe Other names Morton's foot, Greek foot, royal toe, Turkey toe, LaMay toe, Sheppard's toe, Coup d’etoe, Viking toe, Morton's syndrome, [1] long toe, boss toe Dorsal surface of a right foot with Morton's toe (left image) and without (right image). ... Its recorded prevalence varies in different populations, with estimates from 2.95% to 22%. [8] [11] [3] Etymology [ edit ] The name derives from American orthopedic surgeon Dudley Joy Morton (1884–1960), [12] who originally described it as part of Morton's triad (a.k.a. Morton's syndrome or Morton's foot syndrome ): [1] a congenital short first metatarsal bone, a hypermobile first metatarsal segment and calluses under the second and third metatarsals. ... PMID 15324790 . ^ a b Decherchi, Patrick (2005). "Dudley Joy Morton's foot syndrome". Presse Médicale . 34 (22 Pt 1): 1737–40. doi : 10.1016/S0755-4982(05)84262-9 . ... Retrieved 13 November 2016 . ^ Online Mendelian Inheritance in Man: An Online Catalog of Human Genes and Genetic Disorders. http://www.omim.org/entry/189200 ^ Morton's syndrome (Dudley Joy Morton) at Who Named It? ... References [ edit ] Wikimedia Commons has media related to Morton's syndrome . Morton, D. J. "Metatarsus atavicus: the identification of a distinct type of foot disorder", The Journal of Bone and Joint Surgery , Boston, 1927, 9: 531-544.
Overview Morton's neuroma is a painful condition that affects the ball of your foot, most commonly the area between your third and fourth toes. Morton's neuroma may feel as if you are standing on a pebble in your shoe or on a fold in your sock. Morton's neuroma involves a thickening of the tissue around one of the nerves leading to your toes. This can cause a sharp, burning pain in the ball of your foot. You may have stinging, burning or numbness in the affected toes. High-heeled or tight shoes have been linked to the development of Morton's neuroma.
In sleep clinics , obstructive sleep apnea syndrome or obstructive sleep apnea–hypopnea syndrome is normally diagnosed based on the frequent presence of apneas and/or hypopneas rather than differentiating between the two phenomena. ... Daytime hypopnea events, however, are mostly limited to those with severely compromised respiratory muscles , as occurs in certain neuromuscular diseases or compromised central respiratory drive , as occurs in conditions such as acquired or congenital central hypoventilation syndrome (ACHS or CCHS). Daytime hypopnea can also cause a drop in blood oxygen level. ... Causes [ edit ] Among the causes of hypopnea are: anatomical defects such as nasal septum deformation or congenital narrowness of nasal meatus and the gullet acute tonsillitis and/or adenoiditis obesity or being overweight neuromuscular disease or any condition that entails weakened respiratory muscles hypoventilation syndromes involving compromised or failed respiratory drive use of sedatives , for example sleeping pills alcohol abuse smoking aging others, most of which are also typical causes of airway obstruction , snoring and sleep apnea Diagnosis [ edit ] In the context of diagnosis and treatment of sleep disorders , a hypopnea is not considered to be clinically significant unless there is a 30% or greater reduction in flow lasting for 10 seconds or longer and an associated 4% or greater desaturation in the person's O 2 levels , or if it results in arousal or fragmentation of sleep. ... Central hypopnea [ edit ] See also: Central sleep apnea People with neuromuscular disorders or hypoventilation syndromes involving failed respiratory drive experience central hypoventilation. ... book=Medical&va=hypopnea http://www.emedicine.com/neuro/TOPIC419.HTM http://www.sleepdex.org/dyssomnias.htm https://www.sciencedaily.com/releases/2007/10/071015081737.htm http://www.aasmnet.org/Resources/PracticeParameters/PP_MedicalTherapyOSA.pdf v t e Symptoms and signs relating to the respiratory system Auscultation Stethoscope Respiratory sounds Stridor Wheeze Crackles Rhonchi Stertor Squawk Pleural friction rub Fremitus Bronchophony Terminal secretions Elicited findings Percussion Pectoriloquy Whispered pectoriloquy Egophony Breathing Rate Apnea Prematurity Dyspnea Hyperventilation Hypoventilation Hyperpnea Tachypnea Hypopnea Bradypnea Pattern Agonal respiration Biot's respiration Cheyne–Stokes respiration Kussmaul breathing Ataxic respiration Other Respiratory distress Respiratory arrest Orthopnea / Platypnea Trepopnea Aerophagia Asphyxia Breath holding Mouth breathing Snoring Other Chest pain In children Precordial catch syndrome Pleurisy Nail clubbing Cyanosis Cough Sputum Hemoptysis Epistaxis Silhouette sign Post-nasal drip Hiccup COPD Hoover's sign asthma Curschmann's spirals Charcot–Leyden crystals chronic bronchitis Reid index sarcoidosis Kveim test pulmonary embolism Hampton hump Westermark sign pulmonary edema Kerley lines Hamman's sign Golden S sign