-
Carney Complex
Wikipedia
Carney complex Other names LAMB syndrome Specialty Oncology , cardiology Carney complex and its subsets LAMB syndrome [1] and NAME syndrome [1] are autosomal dominant conditions comprising myxomas of the heart and skin, hyperpigmentation of the skin ( lentiginosis ), and endocrine overactivity. [2] [3] It is distinct from Carney's triad . ... Endocrine tumors may manifest as disorders such as Cushing syndrome . The most common endocrine gland manifestation is an ACTH-independent Cushing's syndrome due to primary pigmented nodular adrenocortical disease (PPNAD). The LAMB acronym refers to lentigines , atrial myxomas , and blue nevi . [1] NAME refers to nevi , atrial myxoma, myxoid neurofibromas, and ephelides . [1] Testicular cancer, particularly Sertoli cell type, is associated with Carney syndrome. [5] Thyroid and pancreas cancer may also occur. [6] [7] Although J Aidan Carney also described Carney's triad it is entirely different. [8] Pathophysiology [ edit ] Carney complex is most commonly caused by mutations in the PRKAR1A gene on chromosome 17 (17q23-q24) [9] which may function as a tumor-suppressor gene . ... See also [ edit ] Epithelioid blue nevus List of cutaneous neoplasms associated with systemic syndromes References [ edit ] ^ a b c d Carney Syndrome at eMedicine ^ Carney, J.; Gordon, H.; Carpenter, P.; Shenoy, B.; Go, V. (1985). ... J Endocr Soc 1(10):1312-1321. doi: 10.1210/js.2017-00283 External links [ edit ] Classification D OMIM : 160980 605244 MeSH : D056733 External resources eMedicine : med/2941 Orphanet : 1359 GeneReview/UW/NIH entry on Carney complex v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteins
-
Neurofibromatosis Type Ii
Wikipedia
Type of neurofibromatosis disease Neurofibromatosis type II Other names multiple inherited schwannomas, meningiomas, and ependymomas (MISME syndrome)) Micrograph of a schwannoma , a tumor seen in neurofibromatosis type II. ... Specialty Medical genetics , neurology Neurofibromatosis type II (also known as MISME syndrome – multiple inherited schwannomas, meningiomas, and ependymomas) is a genetic condition which may be inherited or may arise spontaneously. ... External links [ edit ] Classification D ICD - 10 : D33 , Q85.0 ( ILDS Q85.020) ICD - 9-CM : 237.72 OMIM : 101000 MeSH : D016518 DiseasesDB : 8960 External resources MedlinePlus : 000795 eMedicine : neuro/496 radio/475 GeneReviews : Neurofibromatosis 2 v t e Phakomatosis Angiomatosis Sturge–Weber syndrome Von Hippel–Lindau disease Hamartoma Tuberous sclerosis Hypothalamic hamartoma ( Pallister–Hall syndrome ) Multiple hamartoma syndrome Proteus syndrome Cowden syndrome Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Neurofibromatosis Type I Type II Other Abdallat–Davis–Farrage syndrome Ataxia telangiectasia Incontinentia pigmenti Peutz–Jeghers syndrome Encephalocraniocutaneous lipomatosis v t e Tumours of the nervous system Endocrine Sellar : Craniopharyngioma Pituicytoma Other: Pinealoma CNS Neuroepithelial ( brain tumors , spinal tumors ) Glioma Astrocyte Astrocytoma Pilocytic astrocytoma Pleomorphic xanthoastrocytoma Subependymal giant cell astrocytoma Fibrillary astrocytoma Anaplastic astrocytoma Glioblastoma multiforme Oligodendrocyte Oligodendroglioma Anaplastic oligodendroglioma Ependyma Ependymoma Subependymoma Choroid plexus Choroid plexus tumor Choroid plexus papilloma Choroid plexus carcinoma Multiple/unknown Oligoastrocytoma Gliomatosis cerebri Gliosarcoma Mature neuron Ganglioneuroma : Ganglioglioma Retinoblastoma Neurocytoma Dysembryoplastic neuroepithelial tumour Lhermitte–Duclos disease PNET Neuroblastoma Esthesioneuroblastoma Ganglioneuroblastoma Medulloblastoma Atypical teratoid rhabdoid tumor Primitive Medulloepithelioma Meninges Meningioma Hemangiopericytoma Hematopoietic Primary central nervous system lymphoma PNS : Nerve sheath tumor Cranial and paraspinal nerves Neurofibroma Neurofibromatosis Neurilemmoma / Schwannoma Acoustic neuroma Malignant peripheral nerve sheath tumor Other WHO classification of the tumors of the central nervous system Note: Not all brain tumors are of nervous tissue, and not all nervous tissue tumors are in the brain (see brain metastasis ). v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteinsNF2, NF1, SMARCB1, LZTR1, EZR, ACTB, CDKN2A, BAP1, VEGFA, RAC1, YAP1, PAK1, TP53, NEFH, EPB41L3, ETV5, PTGS2, PIK3CA, PGR, MIR296, CD44, CXCR4, MSN, COX8A, MDM2, CTNNB1, MIB1, TCHP, DNMT1, IL17RA, TAZ, AMOT, MIR152, VHL, SAV1, AJUBA, TIMP3, SYK, CADM1, STAT3, SST, SRC, SMS, SMARCE1, IGLL5, SGK1, SRSF5, GOPC, SLC17A6, VTN, FBLIM1, CABIN1, WWC1, LATS2, SIRT2, MORC2, RASSF1, POSTN, PDLIM5, SEMA3F, DCAF1, MSC, HGS, SOCS2, SHANK3, EIF3C, TEAD2, RSS, AGO2, BIN2, BMS1P20, PRKAR1A, RDX, ELF4, CCN1, HTC2, HARS1, MTOR, FLT1, FASN, ESR1, ERBB2, EPO, EPHA1, ENG, EGFR, IL1B, EGF, ATN1, DNMT3A, CYP19A1, CCN2, CRYBB3, CRYBB2, CAPN5, BRCA1, BCR, BCL2, IGLV@, IL10, PXN, PEBP1, PTPRJ, PTGDS, MAP2K2, MAP2K1, AREG, PLG, PLAT, PIK3CG, PIK3CD, PIK3CB, SERPINE1, NHS, KDR, NEDD8, COX2, MTAP, MN1, MKI67, MEN1, MEFV, LRP6, LIMK2, LIMK1, LIF, MTCO2P12

-
Breast Cancer
Medlineplus
A significantly increased risk of breast cancer is also a feature of several rare genetic syndromes. These include Cowden syndrome, which is most often caused by mutations in the PTEN gene; hereditary diffuse gastric cancer, which results from mutations in the CDH1 gene; Li-Fraumeni syndrome, which is usually caused by mutations in the TP53 gene; and Peutz-Jeghers syndrome, which typically results from mutations in the STK11 gene. ... Although breast cancer is more common in women than in men, the mutated gene can be inherited from either the mother or the father. In the other syndromes discussed above, the gene mutations that increase cancer risk also have an autosomal dominant pattern of inheritance.
-
X-Linked Myotubular Myopathy
Wikipedia
External links [ edit ] GeneReview/NCBI/NIH/UW entry on X-Linked Myotubular Myopathy Classification D ICD - 10 : G71.2 OMIM : 310400 MeSH : C538647 External resources Orphanet : 596 v t e Diseases of muscle , neuromuscular junction , and neuromuscular disease Neuromuscular- junction disease autoimmune Myasthenia gravis Lambert–Eaton myasthenic syndrome Neuromyotonia Myopathy Muscular dystrophy ( DAPC ) AD Limb-girdle muscular dystrophy 1 Oculopharyngeal Facioscapulohumeral Myotonic Distal (most) AR Calpainopathy Limb-girdle muscular dystrophy 2 Congenital Fukuyama Ullrich Walker–Warburg XR dystrophin Becker's Duchenne Emery–Dreifuss Other structural collagen disease Bethlem myopathy PTP disease X-linked MTM adaptor protein disease BIN1-linked centronuclear myopathy cytoskeleton disease Nemaline myopathy Zaspopathy Channelopathy Myotonia Myotonia congenita Thomsen disease Neuromyotonia / Isaacs syndrome Paramyotonia congenita Periodic paralysis Hypokalemic Thyrotoxic Hyperkalemic Other Central core disease Mitochondrial myopathy MELAS MERRF KSS PEO General Inflammatory myopathy Congenital myopathy v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteinsMTM1, DNM2, BIN1, RYR1, ACTA1, ORAI1, MTMR14, SELENON, TPM3, TPM2, STIM1, CCDC78, MYH7, MTMR2, DES, CMTM1, MTMR1, PTPRU, ATP2B3, GABRA3, MTMR12, SLC25A3, MAMLD1, REG1A, VIM, IDS, MBNL1, NEFL, ACHE

-
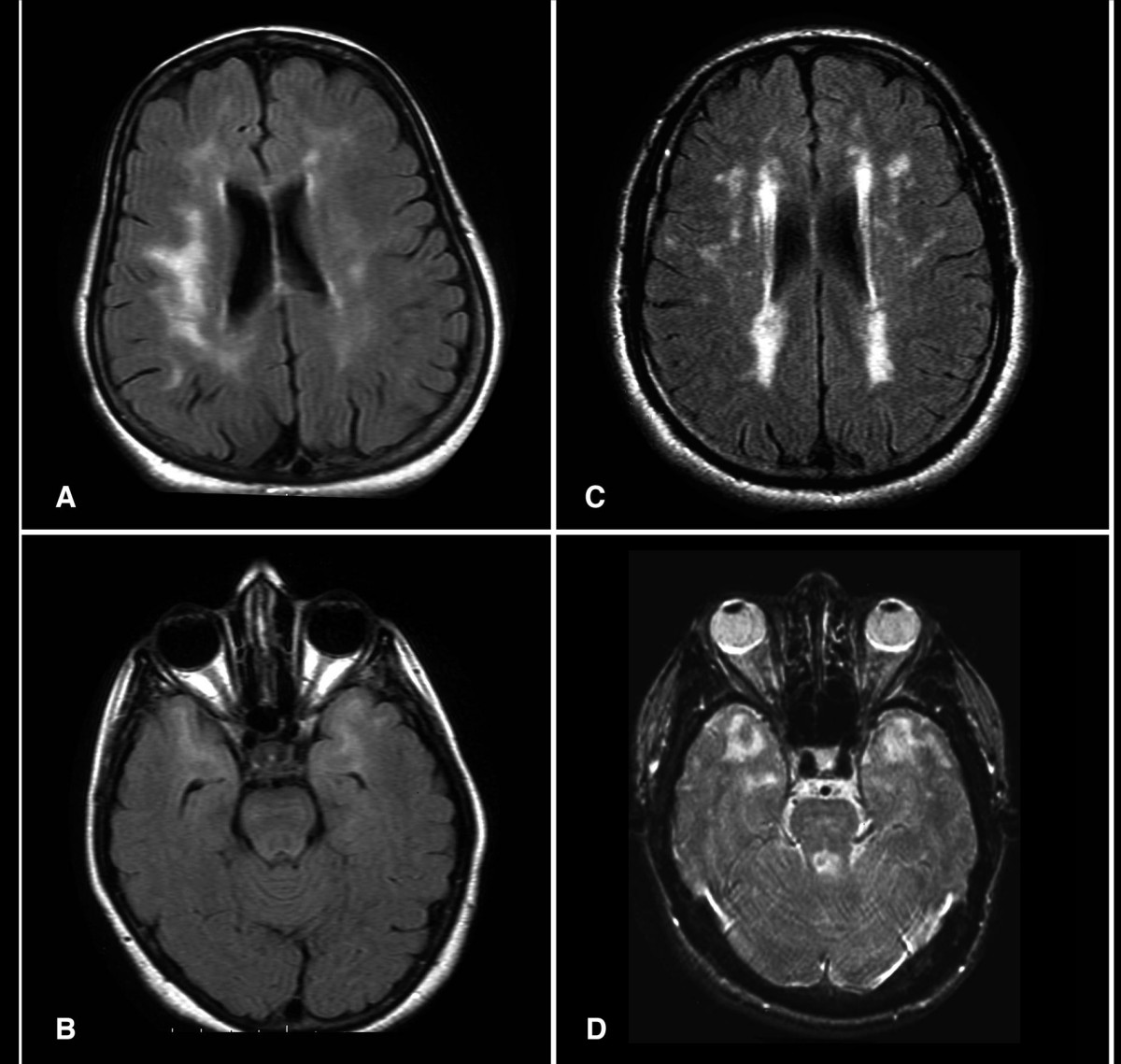
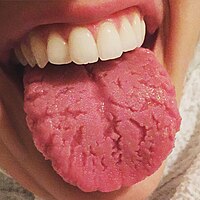
Fissured Tongue
Wikipedia
Associated conditions [ edit ] Fissured tongue is seen in Melkersson-Rosenthal syndrome (along with facial nerve paralysis and granulomatous cheilitis ). It is also seen in most patients with Down syndrome , in association with geographic tongue , in patients with oral manifestations of psoriasis , and in healthy individuals. Fissured tongue is also sometimes a feature of Cowden's syndrome . Cause [ edit ] The cause is unknown, but it may be partly a genetic trait. ... Frequency of Tongue Anomalies in Primary School Of Lahidjan. 3. 2003; 12 (45) :36-42 ] ^ a b Geriatric Nutrition: The Health Professional's Handbook, Ronni Chernoff, (Jones & Bartlett Learning, 2006), page 176 External links [ edit ] Classification D ICD - 10 : K14.5 ICD - 9-CM : 529.5 , 750.13 OMIM : 137400 MeSH : D014063 DiseasesDB : 32503 External resources eMedicine : derm/665 v t e Oral and maxillofacial pathology Lips Cheilitis Actinic Angular Plasma cell Cleft lip Congenital lip pit Eclabium Herpes labialis Macrocheilia Microcheilia Nasolabial cyst Sun poisoning Trumpeter's wart Tongue Ankyloglossia Black hairy tongue Caviar tongue Crenated tongue Cunnilingus tongue Fissured tongue Foliate papillitis Glossitis Geographic tongue Median rhomboid glossitis Transient lingual papillitis Glossoptosis Hypoglossia Lingual thyroid Macroglossia Microglossia Rhabdomyoma Palate Bednar's aphthae Cleft palate High-arched palate Palatal cysts of the newborn Inflammatory papillary hyperplasia Stomatitis nicotina Torus palatinus Oral mucosa – Lining of mouth Amalgam tattoo Angina bullosa haemorrhagica Behçet's disease Bohn's nodules Burning mouth syndrome Candidiasis Condyloma acuminatum Darier's disease Epulis fissuratum Erythema multiforme Erythroplakia Fibroma Giant-cell Focal epithelial hyperplasia Fordyce spots Hairy leukoplakia Hand, foot and mouth disease Hereditary benign intraepithelial dyskeratosis Herpangina Herpes zoster Intraoral dental sinus Leukoedema Leukoplakia Lichen planus Linea alba Lupus erythematosus Melanocytic nevus Melanocytic oral lesion Molluscum contagiosum Morsicatio buccarum Oral cancer Benign: Squamous cell papilloma Keratoacanthoma Malignant: Adenosquamous carcinoma Basaloid squamous carcinoma Mucosal melanoma Spindle cell carcinoma Squamous cell carcinoma Verrucous carcinoma Oral florid papillomatosis Oral melanosis Smoker's melanosis Pemphigoid Benign mucous membrane Pemphigus Plasmoacanthoma Stomatitis Aphthous Denture-related Herpetic Smokeless tobacco keratosis Submucous fibrosis Ulceration Riga–Fede disease Verruca vulgaris Verruciform xanthoma White sponge nevus Teeth ( pulp , dentin , enamel ) Amelogenesis imperfecta Ankylosis Anodontia Caries Early childhood caries Concrescence Failure of eruption of teeth Dens evaginatus Talon cusp Dentin dysplasia Dentin hypersensitivity Dentinogenesis imperfecta Dilaceration Discoloration Ectopic enamel Enamel hypocalcification Enamel hypoplasia Turner's hypoplasia Enamel pearl Fluorosis Fusion Gemination Hyperdontia Hypodontia Maxillary lateral incisor agenesis Impaction Wisdom tooth impaction Macrodontia Meth mouth Microdontia Odontogenic tumors Keratocystic odontogenic tumour Odontoma Dens in dente Open contact Premature eruption Neonatal teeth Pulp calcification Pulp stone Pulp canal obliteration Pulp necrosis Pulp polyp Pulpitis Regional odontodysplasia Resorption Shovel-shaped incisors Supernumerary root Taurodontism Trauma Avulsion Cracked tooth syndrome Vertical root fracture Occlusal Tooth loss Edentulism Tooth wear Abrasion Abfraction Acid erosion Attrition Periodontium ( gingiva , periodontal ligament , cementum , alveolus ) – Gums and tooth-supporting structures Cementicle Cementoblastoma Gigantiform Cementoma Eruption cyst Epulis Pyogenic granuloma Congenital epulis Gingival enlargement Gingival cyst of the adult Gingival cyst of the newborn Gingivitis Desquamative Granulomatous Plasma cell Hereditary gingival fibromatosis Hypercementosis Hypocementosis Linear gingival erythema Necrotizing periodontal diseases Acute necrotizing ulcerative gingivitis Pericoronitis Peri-implantitis Periodontal abscess Periodontal trauma Periodontitis Aggressive As a manifestation of systemic disease Chronic Perio-endo lesion Teething Periapical, mandibular and maxillary hard tissues – Bones of jaws Agnathia Alveolar osteitis Buccal exostosis Cherubism Idiopathic osteosclerosis Mandibular fracture Microgenia Micrognathia Intraosseous cysts Odontogenic : periapical Dentigerous Buccal bifurcation Lateral periodontal Globulomaxillary Calcifying odontogenic Glandular odontogenic Non-odontogenic: Nasopalatine duct Median mandibular Median palatal Traumatic bone Osteoma Osteomyelitis Osteonecrosis Bisphosphonate-associated Neuralgia-inducing cavitational osteonecrosis Osteoradionecrosis Osteoporotic bone marrow defect Paget's disease of bone Periapical abscess Phoenix abscess Periapical periodontitis Stafne defect Torus mandibularis Temporomandibular joints , muscles of mastication and malocclusions – Jaw joints, chewing muscles and bite abnormalities Bruxism Condylar resorption Mandibular dislocation Malocclusion Crossbite Open bite Overbite Overeruption Overjet Prognathia Retrognathia Scissor bite Maxillary hypoplasia Temporomandibular joint dysfunction Salivary glands Benign lymphoepithelial lesion Ectopic salivary gland tissue Frey's syndrome HIV salivary gland disease Necrotizing sialometaplasia Mucocele Ranula Pneumoparotitis Salivary duct stricture Salivary gland aplasia Salivary gland atresia Salivary gland diverticulum Salivary gland fistula Salivary gland hyperplasia Salivary gland hypoplasia Salivary gland neoplasms Benign: Basal cell adenoma Canalicular adenoma Ductal papilloma Monomorphic adenoma Myoepithelioma Oncocytoma Papillary cystadenoma lymphomatosum Pleomorphic adenoma Sebaceous adenoma Malignant: Acinic cell carcinoma Adenocarcinoma Adenoid cystic carcinoma Carcinoma ex pleomorphic adenoma Lymphoma Mucoepidermoid carcinoma Sclerosing polycystic adenosis Sialadenitis Parotitis Chronic sclerosing sialadenitis Sialectasis Sialocele Sialodochitis Sialosis Sialolithiasis Sjögren's syndrome Orofacial soft tissues – Soft tissues around the mouth Actinomycosis Angioedema Basal cell carcinoma Cutaneous sinus of dental origin Cystic hygroma Gnathophyma Ludwig's angina Macrostomia Melkersson–Rosenthal syndrome Microstomia Noma Oral Crohn's disease Orofacial granulomatosis Perioral dermatitis Pyostomatitis vegetans Other Eagle syndrome Hemifacial hypertrophy Facial hemiatrophy Oral manifestations of systemic diseaseKRT6A, AKT1, CC2D2A, WDPCP, TMEM216, MKS1, CEP55, SLC39A4, RPGRIP1, TMEM231, RAB3GAP2, CSPP1, TCTN2, CEP290, B9D2, TMEM107, TMEM67, B9D1, RPGRIP1L, GJB2, SDHD, PIK3CA, PKP1, PTEN, SDHB, RAB3GAP1, SDHC, KDM5C, ECEL1, ABCC9, SEC23B, GJB6, KLLN, KRT16, HLA-DRB1, HLA-C
-
Oguchi Disease
Wikipedia
External links [ edit ] Oguchi disease at NIH 's Office of Rare Diseases Classification D ICD - 9-CM : 368.61 OMIM : 258100 MeSH : C537743 External resources Orphanet : 75382 v t e Diseases of the human eye Adnexa Eyelid Inflammation Stye Chalazion Blepharitis Entropion Ectropion Lagophthalmos Blepharochalasis Ptosis Blepharophimosis Xanthelasma Ankyloblepharon Eyelash Trichiasis Madarosis Lacrimal apparatus Dacryoadenitis Epiphora Dacryocystitis Xerophthalmia Orbit Exophthalmos Enophthalmos Orbital cellulitis Orbital lymphoma Periorbital cellulitis Conjunctiva Conjunctivitis allergic Pterygium Pseudopterygium Pinguecula Subconjunctival hemorrhage Globe Fibrous tunic Sclera Scleritis Episcleritis Cornea Keratitis herpetic acanthamoebic fungal Exposure Photokeratitis Corneal ulcer Thygeson's superficial punctate keratopathy Corneal dystrophy Fuchs' Meesmann Corneal ectasia Keratoconus Pellucid marginal degeneration Keratoglobus Terrien's marginal degeneration Post-LASIK ectasia Keratoconjunctivitis sicca Corneal opacity Corneal neovascularization Kayser–Fleischer ring Haab's striae Arcus senilis Band keratopathy Vascular tunic Iris Ciliary body Uveitis Intermediate uveitis Hyphema Rubeosis iridis Persistent pupillary membrane Iridodialysis Synechia Choroid Choroideremia Choroiditis Chorioretinitis Lens Cataract Congenital cataract Childhood cataract Aphakia Ectopia lentis Retina Retinitis Chorioretinitis Cytomegalovirus retinitis Retinal detachment Retinoschisis Ocular ischemic syndrome / Central retinal vein occlusion Central retinal artery occlusion Branch retinal artery occlusion Retinopathy diabetic hypertensive Purtscher's of prematurity Bietti's crystalline dystrophy Coats' disease Sickle cell Macular degeneration Retinitis pigmentosa Retinal haemorrhage Central serous retinopathy Macular edema Epiretinal membrane (Macular pucker) Vitelliform macular dystrophy Leber's congenital amaurosis Birdshot chorioretinopathy Other Glaucoma / Ocular hypertension / Primary juvenile glaucoma Floater Leber's hereditary optic neuropathy Red eye Globe rupture Keratomycosis Phthisis bulbi Persistent fetal vasculature / Persistent hyperplastic primary vitreous Persistent tunica vasculosa lentis Familial exudative vitreoretinopathy Pathways Optic nerve Optic disc Optic neuritis optic papillitis Papilledema Foster Kennedy syndrome Optic atrophy Optic disc drusen Optic neuropathy Ischemic anterior (AION) posterior (PION) Kjer's Leber's hereditary Toxic and nutritional Strabismus Extraocular muscles Binocular vision Accommodation Paralytic strabismus Ophthalmoparesis Chronic progressive external ophthalmoplegia Kearns–Sayre syndrome palsies Oculomotor (III) Fourth-nerve (IV) Sixth-nerve (VI) Other strabismus Esotropia / Exotropia Hypertropia Heterophoria Esophoria Exophoria Cyclotropia Brown's syndrome Duane syndrome Other binocular Conjugate gaze palsy Convergence insufficiency Internuclear ophthalmoplegia One and a half syndrome Refraction Refractive error Hyperopia Myopia Astigmatism Anisometropia / Aniseikonia Presbyopia Vision disorders Blindness Amblyopia Leber's congenital amaurosis Diplopia Scotoma Color blindness Achromatopsia Dichromacy Monochromacy Nyctalopia Oguchi disease Blindness / Vision loss / Visual impairment Anopsia Hemianopsia binasal bitemporal homonymous Quadrantanopia subjective Asthenopia Hemeralopia Photophobia Scintillating scotoma Pupil Anisocoria Argyll Robertson pupil Marcus Gunn pupil Adie syndrome Miosis Mydriasis Cycloplegia Parinaud's syndrome Other Nystagmus Childhood blindness Infections Trachoma Onchocerciasis v t e Cell membrane protein disorders (other than Cell surface receptor , enzymes , and cytoskeleton ) Arrestin Oguchi disease 1 Myelin Pelizaeus–Merzbacher disease Dejerine–Sottas disease Charcot–Marie–Tooth disease 1B, 2J Pulmonary surfactant Surfactant metabolism dysfunction 1, 2 Cell adhesion molecule IgSF CAM : OFC7 Cadherin : DSG1 Striate palmoplantar keratoderma 1 DSG2 Arrhythmogenic right ventricular dysplasia 10 DSG4 LAH1 DSC2 Arrhythmogenic right ventricular dysplasia 11 Integrin : cell surface receptor deficiencies Tetraspanin TSPAN7 X-Linked mental retardation 58 TSPAN12 Familial exudative vitreoretinopathy 5 Other KIND1 Kindler syndrome HFE HFE hereditary haemochromatosis DYSF Distal muscular dystrophy Limb-girdle muscular dystrophy 2B See also other cell membrane proteins v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteins

-
Pseudopseudohypoparathyroidism
Wikipedia
"Pseudohypoparathyroidism: one gene, several syndromes". Journal of Endocrinological Investigation . 40 (4): 347–356. doi : 10.1007/s40618-016-0588-4 . ... Sadaf Farooqi; Stephen O'Rahilly (12 September 2008). The genetics of obesity syndromes . Oxford University Press US. pp. 91–. ... External links [ edit ] Classification D ICD - 9-CM : 275.49 OMIM : 612463 MeSH : D011556 DiseasesDB : 29783 Look up pseudopseudohypoparathyroidism in Wiktionary, the free dictionary. v t e Parathyroid disease Hypoparathyroidism Pseudohypoparathyroidism Pseudopseudohypoparathyroidism Hyperparathyroidism Primary Secondary Tertiary Osteitis fibrosa cystica Other Parathyroiditis v t e Disorders due to genomic imprinting Chromosome 15 Angelman syndrome ♀ / Prader-Willi syndrome ♂ Chromosome 11 Beckwith–Wiedemann syndrome ♀ / Silver–Russell syndrome ♂ Myoclonic dystonia Chromosome 20 Pseudohypoparathyroidism ♀ / Pseudopseudohypoparathyroidism ♂ Chromosome 6 Transient neonatal diabetes mellitus v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteins
-
Costello Syndrome
Wikipedia
Costello syndrome Other names Faciocutaneoskeletal syndrome Costello syndrome is inherited in an autosomal dominant manner. Specialty Medical genetics Costello syndrome , also called faciocutaneoskeletal syndrome or FCS syndrome , is a rare genetic disorder that affects many parts of the body. ... After his presentation, members of the Costello Syndrome Family Network discussed the possibility of FTIs helping children with Costello syndrome. ... When this was reported in mainstream news, the Costello Syndrome Professional Advisory Board was asked about its use in Costello Syndrome. ... S2CID 22119631 . ^ Lisa Schoyer, 2007 Costello syndrome medical symposium. Some text in this article was originally taken from http://ghr.nlm.nih.gov/condition=costellosyndrome , a public domain source External links [ edit ] Classification D OMIM : 218040 MeSH : D056685 DiseasesDB : 32846 External resources Orphanet : 2143 GeneReviews: Costello Syndrome GeneReviews: Daisy's battle with Costello Syndrome v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteins

-
Aarskog–scott Syndrome
Wikipedia
Aarskog–Scott syndrome / Aarskog Syndrome Other names Faciodigitogenital syndrome (FGDY), faciogenital dysplasia, Aarskog disease, Scott Aarskog syndrome [1] Specialty Medical genetics Symptoms Broad hands and feet, wide set eyes, low set ears, drooping lower lip [1] Causes Genetic ( X-linked recessive ) [1] Deaths 2018, two deaths one patient aged 66 years, another aged 62 also diagnosed with Non-Hodgkin lymphoma 2019 one death aged 54. ... The cause of Aarskog-Scott syndrome in other affected individuals is unknown. [3] Pathophysiology [ edit ] The Aarskog–Scott syndrome is due to mutation in the FGD1 gene. ... Scott, Jr., an American medical geneticist who independently described the syndrome in 1971. [9] References [ edit ] ^ a b c "Aarskog syndrome" . rarediseases.info.nih.gov . ... External links [ edit ] Aarskog–Scott syndrome , detailed up-to-date information in OMIM (Online Mendelian Inheritance in Man) Aarskog's syndrome at Who Named It? The Aarskog Foundation Classification D ICD - 10 : Q87.1 ICD - 9-CM : 759.89 OMIM : 100050 MeSH : C535331 DiseasesDB : 29329 External resources MedlinePlus : 001654 Orphanet : 915 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteins
-
Sturge–weber Syndrome
Wikipedia
Sturge–Weber syndrome Other names Sturge–Weber–Krabbe disease CT scan of Sturge-Weber syndrome Specialty Medical genetics Sturge–Weber syndrome , sometimes referred to as encephalotrigeminal angiomatosis , is a rare congenital neurological and skin disorder. ... More research is needed on this type of Sturge–Weber syndrome. Type 3 has leptomeningeal angioma involvement exclusively. ... "Sturge–Weber Syndrome and Port-Wine Stains Caused by Somatic Mutation in" . ... Further reading [ edit ] Wikimedia Commons has media related to Sturge–Weber syndrome . Greenwood M, Meechan JG (July 2003). ... Fig. 2 A patient with Sturge Weber Syndrome External links [ edit ] Classification D ICD - 10 : Q85.8 ICD - 9-CM : 759.6 OMIM : 185300 MeSH : D013341 DiseasesDB : 12572 External resources MedlinePlus : 001426 eMedicine : neuro/356 Orphanet : 3205 sturge_weber at NINDS v t e Phakomatosis Angiomatosis Sturge–Weber syndrome Von Hippel–Lindau disease Hamartoma Tuberous sclerosis Hypothalamic hamartoma ( Pallister–Hall syndrome ) Multiple hamartoma syndrome Proteus syndrome Cowden syndrome Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Neurofibromatosis Type I Type II Other Abdallat–Davis–Farrage syndrome Ataxia telangiectasia Incontinentia pigmenti Peutz–Jeghers syndrome Encephalocraniocutaneous lipomatosisGNAQ, RASA1, FN1, PNPLA6, LIFR, ALDH18A1, PIK3CG, POSTN, SNAP29, VEGFA, TSC2, TSC1, TEK, PTPRC, ACHE, PIK3CD, CYP1B1, PIK3CA, NF1, MEF2C, KDR, HIF1A, EDN1, CYP2B6, PIK3CB

-
X-Linked Lymphoproliferative Disease
Wikipedia
X-linked lymphoproliferative disease Other names Duncan's disease Specialty Hematology X-linked lymphoproliferative disease (also known as Duncan's disease [1] : 86 or Purtilo syndrome [2] ) is a lymphoproliferative disorder . [3] Contents 1 Presentation 2 Cause 2.1 XLP1 2.2 XLP2 3 Diagnosis 4 Treatment 5 Eponym 6 References 7 External links Presentation [ edit ] Strangely, in boys with X-linked lymphoproliferative disorder, there is an inability to mount an immune response to the Epstein-Barr virus (EBV), [4] which often leads to death from bone marrow failure, irreversible hepatitis, and malignant lymphoma. ... You can help by adding to it . ( December 2017 ) Eponym [ edit ] It is also known as Duncan Disease, after 6 of 18 males in the Duncan family died of lymphoproliferative disease, including fulminant infectious mononucleosis and lymphoma . [11] It is also called "Purtilo's Syndrome", after Dr. David Theodore Purtilo (1939–1992), a pioneering Pathologist and Immunologist at the American Army Center for Pathology in Washington, who discovered it in the early 1970s. ... Philadelphia: Elsevier Saunders. ^ X-linked Lymphoproliferative Syndrome at Merck Manual of Diagnosis and Therapy Professional Edition ^ Rigaud S, Fondanèche MC, Lambert N, et al. ... "XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome". Nature . 444 (7115): 110–4. Bibcode : 2006Natur.444..110R . doi : 10.1038/nature05257 . ... N Engl J Med 1974; 291:736. ^ https://www.nytimes.com/1992/10/03/us/david-t-purtilo-53-a-specialist-in-disorders-of-the-immune-system.html External links [ edit ] GeneReview/NIH/UW entry on Lymphoproliferative Disease, X-Linked Classification D ICD - 10 : D82.3 OMIM : 308240 300635 MeSH : D008232 DiseasesDB : 3998 External resources eMedicine : med/1370 GeneReviews : Lymphoproliferative Disease, X-Linked Orphanet : 2442 v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia v t e Leukaemias , lymphomas and related disease B cell ( lymphoma , leukemia ) (most CD19 CD20 ) By development/ marker TdT+ ALL ( Precursor B acute lymphoblastic leukemia/lymphoma ) CD5 + naive B cell ( CLL/SLL ) mantle zone ( Mantle cell ) CD22 + Prolymphocytic CD11c+ ( Hairy cell leukemia ) CD79a + germinal center / follicular B cell ( Follicular Burkitt's GCB DLBCL Primary cutaneous follicle center lymphoma ) marginal zone / marginal zone B-cell ( Splenic marginal zone MALT Nodal marginal zone Primary cutaneous marginal zone lymphoma ) RS ( CD15 +, CD30 +) Classic Hodgkin lymphoma ( Nodular sclerosis ) CD20+ ( Nodular lymphocyte predominant Hodgkin lymphoma ) PCDs / PP ( CD38 +/ CD138 +) see immunoproliferative immunoglobulin disorders By infection KSHV ( Primary effusion ) EBV Lymphomatoid granulomatosis Post-transplant lymphoproliferative disorder Classic Hodgkin lymphoma Burkitt's lymphoma HCV Splenic marginal zone lymphoma HIV ( AIDS-related lymphoma ) Helicobacter pylori ( MALT lymphoma ) Cutaneous Diffuse large B-cell lymphoma Intravascular large B-cell lymphoma Primary cutaneous marginal zone lymphoma Primary cutaneous immunocytoma Plasmacytoma Plasmacytosis Primary cutaneous follicle center lymphoma T/NK T cell ( lymphoma , leukemia ) (most CD3 CD4 CD8 ) By development/ marker TdT+ : ALL ( Precursor T acute lymphoblastic leukemia/lymphoma ) prolymphocyte ( Prolymphocytic ) CD30+ ( Anaplastic large-cell lymphoma Lymphomatoid papulosis type A ) Cutaneous MF+variants indolent: Mycosis fungoides Pagetoid reticulosis Granulomatous slack skin aggressive: Sézary disease Adult T-cell leukemia/lymphoma Non-MF CD30 -: Non-mycosis fungoides CD30− cutaneous large T-cell lymphoma Pleomorphic T-cell lymphoma Lymphomatoid papulosis type B CD30 +: CD30+ cutaneous T-cell lymphoma Secondary cutaneous CD30+ large-cell lymphoma Lymphomatoid papulosis type A Other peripheral Hepatosplenic Angioimmunoblastic Enteropathy-associated T-cell lymphoma Peripheral T-cell lymphoma not otherwise specified ( Lennert lymphoma ) Subcutaneous T-cell lymphoma By infection HTLV-1 ( Adult T-cell leukemia/lymphoma ) NK cell / (most CD56 ) Aggressive NK-cell leukemia Blastic NK cell lymphoma T or NK EBV ( Extranodal NK-T-cell lymphoma / Angiocentric lymphoma ) Large granular lymphocytic leukemia Lymphoid+ myeloid Acute biphenotypic leukaemia Lymphocytosis Lymphoproliferative disorders ( X-linked lymphoproliferative disease Autoimmune lymphoproliferative syndrome ) Leukemoid reaction Diffuse infiltrative lymphocytosis syndrome Cutaneous lymphoid hyperplasia Cutaneous lymphoid hyperplasia with bandlike and perivascular patterns with nodular pattern Jessner lymphocytic infiltrate of the skin General Hematological malignancy leukemia Lymphoproliferative disorders Lymphoid leukemias v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteinsSH2D1A, IL17A, XIAP, IFNG, STAG2, SLAMF1, APCS, IL10, HPRT1, ITK, RTEL1, CD244, CD1D, ICOS, CCR4, TRBV20OR9-2, NR0B2, CD84, TENM1, HPGDS, IL22, RAB27A, FOXP3, AP5M1, NLRC4, IL21, SLAMF6, VPS13B, CD2, PTPN6, PTPN11, IL2RG, DOK1, FUT1, HLA-DQB1, LYST, IL1A, IL1B, IL1RN, IL4, CYBB, IL7R, CD40LG, INS, CD27, PDCD1, SERPINA4, PLCG1, NCF1
-
Follicular Thyroid Cancer
Wikipedia
Contents 1 Cause 1.1 Associated mutations 2 Hurthle cell variant 3 Diagnosis 3.1 Classification 4 Treatment 4.1 Initial treatment 4.2 Finding disease recurrence 5 Prognosis 6 References 7 External links Cause [ edit ] Associated mutations [ edit ] Approximately one-half of follicular thyroid carcinomas have mutations in the Ras subfamily of oncogenes, most notably HRAS , NRAS , and KRAS . [1] Mutations in MINPP1 have likewise been observed, as well as germline PTEN gene mutations responsible for Cowden syndrome of which follicular thyroid cancer is a feature.THRB, NRAS, HRAS, PTEN, KRAS, DIRAS3, PRKAR1A, PIK3CA, AKT1, SDHD, MINPP1, TP53, TAS2R38, FOXE1, PTCH1, HABP2, PPARG, SRGAP1, CCDC6, PIK3CG, PIK3CD, PIK3CB, F9, RET, PAX8, KLLN, SDHC, SEC23B, SDHB, BRAF, TG, TSHR, TERT, LOC110806263, RASSF1, VEGFA, EGF, MIR146B, DPP4, SLC5A5, BRCA1, KDR, CTNNB1, LGALS3, HIF1A, CD274, NME1, PROX1, FLT1, RAPGEF3, SGSM3, MMP9, RASAL1, SRY, H2AX, CLDN1, IDH1, MMP2, PTGS2, TPCN1, TPT1, YY1, APC, MIR146A, EGFR, CLDN7, MIR21, NKX2-1, TFF3, MIR199B, ST8SIA4, TIE1, THBS1, BUB3, TGFB1, TAF1, STK11, KL, RPH3AL, STC1, SQSTM1, PER2, SPHK1, TNF, TPM3, FCGBP, CDK5R1, VDR, IQGAP1, VEGFC, TNFSF10, MADD, CAVIN2, FZD1, HMGA2, EZR, VIM, GDF15, HCP5, BMS1, KLB, ATF7IP, MIB1, ACE2, LIMD2, COL18A1, RASSF5, MRO, ST6GAL2, HTRA3, PRRT2, NIBAN1, CYP2R1, HT, TICAM1, ANO5, RAB23, TMED10P1, IYD, MIR10B, MIR183, MIR197, MIR19A, MIR221, MIR346, MIR885, MIR4299, HOTAIRM1, H3P9, H3P17, H3P47, DUOX1, ABI3, TSHZ1, RAPGEF4, C1D, CIB1, MRPL28, TXNRD2, ST6GALNAC2, PDPN, POSTN, SPINT2, KHDRBS1, PTGES3, SUB1, RAB40B, TMED10, OGFR, CORO1A, DCTN4, CHEK2, SMG1, TBC1D9, DICER1, QPRT, NUP62, SOSTDC1, NUPR1, ST6GALNAC4, DLL1, HIPK2, NXT1, DUOX2, ANGPTL4, SST, ACACA, SPINT1, FGF2, DIO1, DIO2, DPEP1, DUSP5, E2F1, ERBB2, ERCC1, ESR2, ETS1, ETV6, EVPL, EZH2, FBN1, FGFR4, CYP27B1, FHIT, FOXO3, FLT3, FOLH1, FOSB, MTOR, GDF10, GJB2, GPC3, GOT2, UTS2R, GPER1, GTF2H1, ACE, CYP24A1, SOD2, CAD, ANPEP, ANXA2, XIAP, APLP2, APP, AQP7, AR, ARNTL, ATM, BNIP3, BSG, BUB1, BUB1B, CAV1, CCN2, CAV2, CCND2, SCARB1, CD44, CD68, CDKN1B, CDKN1C, CDKN2A, CDKN2B, CEACAM5, CGA, CLDN4, CRY2, HGF, HLA-G, HMBS, HTRA1, OGG1, OGN, PA2G4, SERPINE1, PCSK2, ENPP2, ABCB1, PHB, SERPINB5, PLG, PRKCA, PRKCB, MAPK3, PSMB6, NR4A1, ACTN4, PTK2, RAP1A, RAP1B, RARA, RRAS, RRM2, S100A10, SCT, CCL15, CXCL12, SLC2A1, SLC2A3, NTRK1, NTHL1, NOTCH3, NOTCH1, HSPA9, HSP90AA1, TNC, ICAM1, IDH2, IGF1R, IGFBP6, IGFBP7, IL1A, IL1B, IL6, CXCL8, CXCR1, IL13RA2, INS, INSL3, IRAK1, ITGA6, ITGAV, STT3A, BCAM, MKI67, MMP1, MT1G, MT1M, NCAM1, NNMT, H3P10

-
Carpenter Syndrome
Wikipedia
Teeth are usually very late to come in and will be undersized and spaced far apart (Carpenter Syndrome-description). Other physical abnormalities often associated with Carpenter Syndrome include extra digits. ... Genetics [ edit ] Carpenter syndrome has an autosomal recessive pattern of inheritance . ... The testes of males affected by Carpenter Syndrome may also fail to descend (Paul A. ... "Craniofacial dysmorphology of Carpenter syndrome: lessons from three affected siblings". ... PMID 23063620 . ^ a b Paul A. Johnson, 2002 ^ Carpenter Syndrom-What is it?, 2007 External links [ edit ] Classification D ICD - 10 : Q87.0 OMIM : 201000 MeSH : C563187 C563187, C563187 DiseasesDB : 29583 SNOMED CT : 205813009 External resources Orphanet : 65759 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteins
-
Bardet–biedl Syndrome
Wikipedia
"Laurence-Moon-Biedl syndrome" and "Laurence-Moon-Biedl-Bardet" redirect here. Not to be confused with Laurence–Moon syndrome . Bardet–Biedl syndrome Other names Biedl-Bardet Syndrome [1] This condition is often inherited via autosomal recessive manner (including digenic recessive); but epigenetic phennomena also cause some of the variation seen in BBS Specialty Medical genetics Bardet–Biedl syndrome ( BBS ) is a ciliopathic human genetic disorder that produces many effects and affects many body systems. ... Laurence–Moon syndrome is usually considered a separate entity. ... The Clinical, Molecular, and Functional Genetics of Bardet–Biedl Syndrome , in Genetics of Obesity Syndromes . ... External links [ edit ] Classification D ICD - 10 : Q87.8 ICD - 9-CM : 759.89 OMIM : 209900 MeSH : D020788 DiseasesDB : 7286 External resources GeneReviews : Bardet-Biedl Syndrome Orphanet : 110 Overview at United States National Library of Medicine Molecular diagnosis at NCBI v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome v t e Diseases of cilia Structural receptor: Polycystic kidney disease cargo: Asphyxiating thoracic dysplasia basal body : Bardet–Biedl syndrome mitotic spindle : Meckel syndrome centrosome : Joubert syndrome Signaling Nephronophthisis Other/ungrouped Alström syndrome Primary ciliary dyskinesia Senior–Løken syndrome Orofaciodigital syndrome 1 McKusick–Kaufman syndrome Autosomal recessive polycystic kidney See also: ciliary proteins v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteinsMKS1, BBS10, SDCCAG8, LZTFL1, BBIP1, CEP290, ARL6, IFT27, BBS2, MKKS, BBS4, BBS1, TTC8, ALMS1, IFT172, C8orf37, NPHP1, TMEM67, TBC1D32, BBS7, TRIM32, BBS9, BBS5, BBS12, TRAPPC3, ASTN2, PIGX, CEP19, ZDHHC24, CCDC28B, LEP, GLIS2, RTEL1, PLLP, PYY3, SULT1A4, PEG13, AZIN1, STOML3, RIN2, MAGEL2, CBLL2, MARVELD2, MYO3B, INPP5E, USP35, MUL1, WDPCP, IFT74, CEP131, SCAPER, INVS, NPY2R, NPY, NMB, NDN, MYO9A, MYO7A, RAB8A, KIFC3, KIF2A, INSR, IL18, GPT, GFAP, ERG, CCT, TNFRSF11B, PRKN, PCM1, ADIPOQ, CEP164, STUB1, FEM1B, RAPGEF5, IQCB1, FEZ1, TRPV1, PDE6B, VEGFA, SULT1A3, RHO, RFX1, RCVRN, PECAM1, SLX1A-SULT1A3
-
Macrocephaly
Wikipedia
People diagnosed with macrocephaly will have further testing done to determine if the syndrome is accompanied by any other disorders . ... Pathologic macrocephaly is called "syndromic", when it is associated with any other noteworthy condition, and "nonsyndromic" otherwise. Pathologic macrocephaly may be caused by congenital anatomic abnormalities, genetic conditions, or by environmental events. [1] Many genetic conditions are associated with macrocephaly, including familial macrocephaly related to the holgate gene, autism , PTEN mutations such as Cowden disease , neurofibromatosis type 1, and tuberous sclerosis ; overgrowth syndromes such as Sotos syndrome (cerebral gigantism), Weaver syndrome , Simpson-Golabi-Behmel syndrome (bulldog syndrome), and macrocephaly-capillary malformation ( M-CMTC ) syndrome; neurocardiofacial-cutaneous syndromes such as Noonan syndrome , Costello syndrome , Gorlin Syndrome , [2] (also known as Basal Cell Nevus Syndrome) and cardiofaciocutaneous syndrome ; Fragile X syndrome ; leukodystrophies (brain white matter degeneration) such as Alexander disease , Canavan disease , and megalencephalic leukoencephalopathy with subcortical cysts ; and glutaric aciduria type 1 and D-2-hydroxyglutaric aciduria . [1] At one end of the genetic spectrum, duplications of chromosomes have been found to be related to autism and macrocephaly; at the other end, deletions of chromosomes have been found to be related to schizophrenia and microcephaly . [3] [4] [5] Environmental events associated with macrocephaly include infection, neonatal intraventricular hemorrhage (bleeding within the infant brain), subdural hematoma (bleeding beneath the outer lining of the brain), subdural effusion (collection of fluid beneath the outer lining of the brain), and arachnoid cysts (cysts on the brain surface). [1] In research, cranial height or brain imaging may be used to determine intracranial volume more accurately. [1] Below is a list of causes of macrocephaly from Swaiman's pediatric neurology: principles and practice noted in The Little Black Book of Neurology : [6] [7] Hydrocephalus [ edit ] Dandy-Walker malformation Noncommunicating [ edit ] Arnold-Chiari malformation Aqueductal stenosis X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS) syndrome ( L1CAM ) Dandy-Walker malformation Galenic vein aneurysm or malformation Neoplasms , supratentorial , and infratentorial Arachnoid cyst , infratentorial Holoprosencephaly with dorsal interhemispheric sac Communicating [ edit ] External or extraventricular obstructive hydrocephalus ( dilated subarachnoid space) Arachnoid cyst, supratentorial [ edit ] This section is empty. ... Symptoms vary on the cause of macrocephaly on the child and if the child has any other accompanying syndromes which will be determined through molecular testing. ... Treatment [ edit ] Treatment varies depending on whether or not it occurs with other medical conditions in the child and where cerebrospinal fluid is present. [9] If benign and found between the brain and skull then no surgery is needed. [9] [11] If excess fluid is found between the ventricle spaces in the brain then surgery will be needed. [11] Associated syndromes [ edit ] Below is a list of syndromes associated with macrocephaly that are noted in Signs and Symptoms of Genetic Conditions: A Handbook . [12] Lujan-fryns syndrome Include multiple major and or minor anomalies [ edit ] Acrocallosal Syndrome Apert Syndrome Bannayan-Riley-Ruvalcaba Cardiofaciocutaneous syndrome Chromosome 14 - maternal dismoy Chromosome 22qter deletion Cleidocranial dysostosis Costello syndrome Encephalocraniocutaneous lipomatosis FG syndrome Hailermann-streiff syndrome Hydolethalus syndrome Hypomelanosis syndrome Hypomelanosis of Ito Kelvin Peter anomaly plus syndrome Lujan-fryns syndrome Macrocephaly-CM (MCAP) Marshall-Smith syndrome Neuhauser megalocornea/MR syndrome Neurofibromatosis I Nevoid basal cell carcinoma syndrome Noonan syndrome Ocular-ectodermal syndrome Osteopathia striata - cranial sclerosis Perlman syndrome Ricky Ritscher - schinzel syndrome Robinow syndrome Simpson-golabi-behmel syndrome Sotos syndrome Sturge-Weber syndrome Weaver syndrome Wiedermann-rautenstrauch syndrome Sturge-Weber syndrome Secondary to a metabolic disorder [ edit ] Glutaric aciduria type II GM1 gangliosidosis Hunter syndrome Hurler syndrome MPS VII Sanfilippo syndrome Zellweger syndrome Alexander disease Associated with a skeletal dysplasia [ edit ] Achondroplasia Camptomelic dysplasia Craniodiaphyseal dysplasia Craniometalphyseal dysplasia Hypochondrogenesis Hypochondroplasia Kenny-caffey Kniest syndrome Lenz-majewski Osteogenesis imperfecta III Osteopetrosis, autosomal recessive form Schneckenbecken dysplasia Sclerosteosis Short rib syndrome , beemer-langer type Short rib-polydactyly 2 (majewski type) Spondyleopiphyseal dysplasia congenita Thanatophoric dysplasia Tay-sachs With no obvious physical findings [ edit ] Alexander disease Canavan Cobalamin deficiency (combined methylmalonic aciduria and homocystinuria ) Dandy-Walker malformation Glutaric aciduria I L-2 hydroxglutaric aciduria Megalencephalic leukoencephalopathy Osteogenesis imperfecta IV Osteopathia striata-cranial sclerosis Periventricular heterotopia Sandhoff disease Tay-sachs See also [ edit ] Microcephaly Megalencephaly Hydrocephalus References [ edit ] ^ a b c d Williams CA, Dagli A, Battaglia A (2008).

-
Birt–hogg–dubé Syndrome
Wikipedia
Rare autosomal dominant cancer syndrome Birt–Hogg–Dubé syndrome The characteristic fibrofolliculomas of Birt–Hogg–Dubé syndrome seen on a person's face. Specialty Medical genetics Birt–Hogg–Dubé syndrome ( BHD ), also Hornstein–Birt–Hogg–Dubé syndrome , Hornstein–Knickenberg syndrome , and fibrofolliculomas with trichodiscomas and acrochordons [1] is a human autosomal dominant genetic disorder that can cause susceptibility to kidney cancer , renal and pulmonary cysts , and noncancerous tumors of the hair follicles , called fibrofolliculomas . ... They are differentiated with examination of the tumors' histology . [5] Hereditary recurrent pneumothorax or pulmonary cysts are associated with Marfan syndrome , Ehlers–Danlos syndrome , tuberous sclerosis complex (TSC), alpha1-antitrypsin deficiency , and cystic fibrosis . ... Other diseases can mimic the dermatologic manifestations of BHD, including tuberous sclerosis complex, Cowden syndrome , familial trichoepitheliomas , and multiple endocrine neoplasia type 1 . [5] Tuberous sclerosis must be distinguished because both disorders can present with angiofibromas on the face, though they are more common in tuberous sclerosis. [6] Management [ edit ] The different manifestations of Birt–Hogg–Dubé syndrome are controlled in different ways. ... Jelle; De Jong, Daphne; Horenblas, Simon; Ryu, Jay (5 October 2009), "Birt-Hogg-Dubé Syndrome" , in Riegert-Johnson, Douglas L; Boardman, Lisa A; Hefferon, Timothy; Roberts, Maegan (eds.), Cancer Syndromes , Bethesda, MD: National Center for Biotechnology Information, PMID 21249760 Riegert-Johnson, DL, "Birt-Hogg-Dube" , Familial Cancer Syndromes , NCBI , retrieved 21 July 2009 Sudarshan, Sunil; Karam, Jose A.; Brugarolas, James; Thompson, R.

-
Cherubism
Wikipedia
Cherubism has also been found combined with other genetic disorders including Noonan syndrome , Ramon syndrome , and fragile X syndrome . [7] Mutations of the SH3BP2 gene are only reported in 75% of Cherubism cases. [2] The mutation of the SH3BP2 gene is believed to increase production of over active proteins from this gene. ... External links [ edit ] Classification D ICD - 10 : K10.8 ICD - 9-CM : 526.89 OMIM : 118400 MeSH : D002636 DiseasesDB : 31217 External resources MedlinePlus : 001234 eMedicine : radio/284 Orphanet : 184 v t e Dental disease involving the jaw General Jaw abnormality malocclusion Orthodontics Gnathitis Size Micrognathism Maxillary hypoplasia Maxilla and Mandible Cherubism Congenital epulis Torus mandibularis Torus palatinus Other Jaw and base of cranium Prognathism Retrognathism Dental arch Crossbite Overbite Temporomandibular joint disorder v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteinsSH3BP2, TNF, NFATC1, CD68, PTPN11, SYK, CA2, SRC, SH3BP5, USP6, PSTPIP2, TNKS, TNFSF11, RAC1, CALCA, PLEK, PLCG2, PCNA, SERPINE1, FGFR3, CTSK, BAMBI
-
Pseudohypoaldosteronism
Wikipedia
TRPV6 may be involved. [2] Presentation [ edit ] PHA2 is clinically characterised by hypertension, hyperkalaemia , metabolic acidosis and normal renal function. [3] Mechanism [ edit ] PHA2 is also known as familial hyperkalaemic hypertension, or Gordon syndrome . The underlying genetic defect leads to increased sodium chloride reabsorption in the distal tubule in the kidney, leading to volume expansion, hypertension and lowered renin levels. ... Alternatively, WNK4 mutations that result in a gain of function of the Na-Cl co-transporter may inhibit ROMK activity resulting in hyperkalemia. [4] Unlike in PHA1 in which aldosterone resistance is present, in PHA2 the volume expansion leads to relatively low aldosterone levels. [3] Treatment [ edit ] Treatment of severe forms of PHA1 requires relatively large amounts of sodium chloride . [5] These conditions also involve hyperkalemia . [6] In contrast, PHA2 (Gordon's syndrome) requires salt restriction and use of thiazide diuretics to block sodium chloride reabsorption and normalise blood pressure and serum potassium. [ citation needed ] History [ edit ] This syndrome was first described by Cheek and Perry in 1958. [7] Later pediatric endocrinologist Aaron Hanukoglu reported that there are two independent forms of PHA with different inheritance patterns: A renal form with autosomal dominant inheritance exhibiting salt loss mainly from the kidneys, and a multi-system form with autosomal recessive form exhibiting salt loss from kidney, lung, and sweat and salivary glands. [8] [9] The hereditary lack of responsiveness to aldosterone could be due to at least two possibilities: 1. ... PMID 20181799 . ^ a b O'Shaughnessy, Kevin M. (November 2015). "Gordon Syndrome: a continuing story". Pediatric Nephrology (Berlin, Germany) . 30 (11): 1903–1908. doi : 10.1007/s00467-014-2956-7 . ... S2CID 9764720 . ^ Pseudohypoaldosteronism at the US National Library of Medicine Medical Subject Headings (MeSH) ^ CHEEK DB, PERRY JW (1958). "A salt wasting syndrome in infancy" . Arch Dis Child . 33 (169): 252–6. doi : 10.1136/adc.33.169.252 . ... External links [ edit ] GeneReviews/NCBI/NIH/UW entry on Pseudohypoaldosteronism Type II Classification D ICD - 10 : N25.8 OMIM : 177735 614495 614491 614496 614492 145260 264350 177735 614495 614491 614496 614492 145260 MeSH : D011546 External resources eMedicine : article/924100 Orphanet : 444916 v t e Adrenal gland disorder Hyperfunction Aldosterone Hyperaldosteronism Primary aldosteronism Conn syndrome Bartter syndrome Glucocorticoid remediable aldosteronism AME Liddle's syndrome 17α CAH Pseudohypoaldosteronism Cortisol Cushing's syndrome Pseudo-Cushing's syndrome Steroid-induced osteoporosis Sex hormones 21α CAH 11β CAH Hypofunction Aldosterone Hypoaldosteronism 21α CAH 11β CAH Cortisol CAH Lipoid 3β 11β 17α 21α Sex hormones 17α CAH Inborn errors of steroid metabolism Adrenal insufficiency Adrenal crisis Adrenalitis Xanthogranulomatous Addison's disease Waterhouse–Friderichsen syndrome v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions ) v t e Diseases of ion channels Calcium channel Voltage-gated CACNA1A Familial hemiplegic migraine 1 Episodic ataxia 2 Spinocerebellar ataxia type-6 CACNA1C Timothy syndrome Brugada syndrome 3 Long QT syndrome 8 CACNA1F Ocular albinism 2 CSNB2A CACNA1S Hypokalemic periodic paralysis 1 Thyrotoxic periodic paralysis 1 CACNB2 Brugada syndrome 4 Ligand gated RYR1 Malignant hyperthermia Central core disease RYR2 CPVT1 ARVD2 Sodium channel Voltage-gated SCN1A Familial hemiplegic migraine 3 GEFS+ 2 Febrile seizure 3A SCN1B Brugada syndrome 6 GEFS+ 1 SCN4A Hypokalemic periodic paralysis 2 Hyperkalemic periodic paralysis Paramyotonia congenita Potassium-aggravated myotonia SCN4B Long QT syndrome 10 SCN5A Brugada syndrome 1 Long QT syndrome 3 SCN9A Erythromelalgia Febrile seizure 3B Paroxysmal extreme pain disorder Congenital insensitivity to pain Constitutively active SCNN1B / SCNN1G Liddle's syndrome SCNN1A / SCNN1B / SCNN1G Pseudohypoaldosteronism 1AR Potassium channel Voltage-gated KCNA1 Episodic ataxia 1 KCNA5 Familial atrial fibrillation 7 KCNC3 Spinocerebellar ataxia type-13 KCNE1 Jervell and Lange-Nielsen syndrome Long QT syndrome 5 KCNE2 Long QT syndrome 6 KCNE3 Brugada syndrome 5 KCNH2 Short QT syndrome KCNQ1 Jervell and Lange-Nielsen syndrome Romano–Ward syndrome Short QT syndrome Long QT syndrome 1 Familial atrial fibrillation 3 KCNQ2 BFNS1 Inward-rectifier KCNJ1 Bartter syndrome 2 KCNJ2 Andersen–Tawil syndrome Long QT syndrome 7 Short QT syndrome KCNJ11 TNDM3 KCNJ18 Thyrotoxic periodic paralysis 2 Chloride channel CFTR Cystic fibrosis Congenital absence of the vas deferens CLCN1 Thomsen disease Myotonia congenita CLCN5 Dent's disease CLCN7 Osteopetrosis A2, B4 BEST1 Vitelliform macular dystrophy CLCNKB Bartter syndrome 3 TRP channel TRPC6 FSGS2 TRPML1 Mucolipidosis type IV Connexin GJA1 Oculodentodigital dysplasia Hallermann–Streiff syndrome Hypoplastic left heart syndrome GJB1 Charcot–Marie–Tooth disease X1 GJB2 Keratitis–ichthyosis–deafness syndrome Ichthyosis hystrix Bart–Pumphrey syndrome Vohwinkel syndrome ) GJB3 / GJB4 Erythrokeratodermia variabilis Progressive symmetric erythrokeratodermia GJB6 Clouston's hidrotic ectodermal dysplasia Porin AQP2 Nephrogenic diabetes insipidus 2 See also: ion channels v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteinsKLHL3, NR3C2, SCNN1A, SCNN1B, SCNN1G, WNK4, WNK1, GNAS, STX16, GNAS-AS1, CUL3, REN, ASIC5, TNF, NR4A1, CLDN8, MRC1, KCNJ1, SGK1
-
Joubert Syndrome
Wikipedia
Joubert syndrome Other names CPD IV [1] Joubert syndrome is inherited via an autosomal recessive manner Specialty Medical genetics Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum , an area of the brain that controls balance and coordination. ... Rarely, Joubert syndrome is inherited in an X-linked recessive pattern. ... The cilia defects adversely affect "numerous critical developmental signaling pathways" essential to cellular development and thus offer a plausible hypothesis for the often multi-symptom nature of a large set of syndromes and diseases. [ citation needed ] Currently recognized ciliopathies include Joubert syndrome, primary ciliary dyskinesia (also known as Kartagener Syndrome), Bardet–Biedl syndrome , polycystic kidney disease and polycystic liver disease , nephronophthisis , Alström syndrome , Meckel–Gruber syndrome and some forms of retinal degeneration . [17] Joubert syndrome type 2 is disproportionately frequent among people of Jewish descent . [18] References [ edit ] ^ RESERVED, INSERM US14-- ALL RIGHTS. ... "Joubert syndrome with associated corpus callosum agenesis". ... External links [ edit ] Classification D ICD - 10 : Q04.3 ICD - 9-CM : 742.2 OMIM : 213300 DiseasesDB : 30688 External resources GeneReviews : Joubert Syndrome and Related Disorders Orphanet : 475 NINDS Joubert Syndrome Information Page Researchers Identify Joubert Syndrome Genes GeneReviews: Joubert syndrome NCBI Joubert Syndrome v t e Diseases of cilia Structural receptor: Polycystic kidney disease cargo: Asphyxiating thoracic dysplasia basal body : Bardet–Biedl syndrome mitotic spindle : Meckel syndrome centrosome : Joubert syndrome Signaling Nephronophthisis Other/ungrouped Alström syndrome Primary ciliary dyskinesia Senior–Løken syndrome Orofaciodigital syndrome 1 McKusick–Kaufman syndrome Autosomal recessive polycystic kidney See also: ciliary proteins v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteinsINPP5E, AHI1, CPLANE1, TMEM67, KIAA0586, MKS1, PIBF1, TCTN1, TCTN2, CSPP1, CEP104, CEP41, ARMC9, TMEM237, HYLS1, ARL13B, RPGRIP1L, KIAA0753, KIAA0556, TMEM231, TMEM216, B9D2, B9D1, KIF7, CEP120, ARL3, TCTN3, FAM149B1, POC1B, TMEM138, ZIC1, CEP290, CC2D2A, OFD1, PDE6D, ITPR1, RCOR1, RNU4ATAC, ALX4, MVK, MEF2C, TFAP2A, IFT172, LRRCC1, C2CD3, NPHP1, LPO, CLUAP1, INVS, ARL13A, NPHP4, TMEM107, WNT1, CELSR2, BARHL1, BPIFA4P, ITFG1, TIPRL, PPP1R21, LCA5, METTL8, RPGRIP1, ANPEP, CHD7, SUFU, ATF4, CCND1, CSF2, TIMM8A, DVL1, FGF8, GYS2, INPP5B, JBS, LAMC2, RAC2, RASA1, RASGRF1, SMO, SOS1, TULP3, ZNF131, MKKS, MLRL, SYNJ1, SYNJ2, HAP1, ADGRG1, RGS6, KAT5, ZNF423, DMXL2, NXPH1, CEND1, LCA10
-
Cadasil
Wikipedia
CADASIL Other names CADASIL syndrome Brain MRI from patients with CADASIL showing multiple lesions. Specialty Neurology , cardiology , medical genetics CADASIL or CADASIL syndrome , involving cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy , is the most common form of hereditary stroke disorder, and is thought to be caused by mutations of the Notch 3 gene on chromosome 19 . [1] The disease belongs to a family of disorders called the leukodystrophies . ... The mean age of onset of ischemic episodes is approximately 46 years (range 30–70). A classic lacunar syndrome occurs in at least two-thirds of affected patients while hemispheric strokes are much less common. ... Further reading [ edit ] Wikimedia Commons has media related to CADASIL syndrome . Lesnik Oberstein SA, Boon EM, Terwindt GM (June 28, 2012). ... External links [ edit ] cureCADASIL (US) [1] CADASIL Support UK [2] asociacion cadasil espana (Spain) [3] United Leukodystrophy Foundation: CADASIL A patient story at The New York Times Classification D ICD - 10 : I67.850 OMIM : 125310 MeSH : D046589 DiseasesDB : 2161 External resources Patient UK : CADASIL GeneReviews : CADASIL Orphanet : 136 v t e Cerebrovascular diseases including stroke Ischaemic stroke Brain Anterior cerebral artery syndrome Middle cerebral artery syndrome Posterior cerebral artery syndrome Amaurosis fugax Moyamoya disease Dejerine–Roussy syndrome Watershed stroke Lacunar stroke Brain stem Brainstem stroke syndrome Medulla Medial medullary syndrome Lateral medullary syndrome Pons Medial pontine syndrome / Foville's Lateral pontine syndrome / Millard-Gubler Midbrain Weber's syndrome Benedikt syndrome Claude's syndrome Cerebellum Cerebellar stroke syndrome Extracranial arteries Carotid artery stenosis precerebral Anterior spinal artery syndrome Vertebrobasilar insufficiency Subclavian steal syndrome Classification Brain ischemia Cerebral infarction Classification Transient ischemic attack Total anterior circulation infarct Partial anterior circulation infarct Other CADASIL Binswanger's disease Transient global amnesia Haemorrhagic stroke Extra-axial Epidural Subdural Subarachnoid Cerebral/Intra-axial Intraventricular Brainstem Duret haemorrhages General Intracranial hemorrhage Aneurysm Intracranial aneurysm Charcot–Bouchard aneurysm Other Cerebral vasculitis Cerebral venous sinus thrombosis v t e Deficiencies of intracellular signaling peptides and proteins GTP-binding protein regulators GTPase-activating protein Neurofibromatosis type I Watson syndrome Tuberous sclerosis Guanine nucleotide exchange factor Marinesco–Sjögren syndrome Aarskog–Scott syndrome Juvenile primary lateral sclerosis X-Linked mental retardation 1 G protein Heterotrimeic cAMP / GNAS1 : Pseudopseudohypoparathyroidism Progressive osseous heteroplasia Pseudohypoparathyroidism Albright's hereditary osteodystrophy McCune–Albright syndrome CGL 2 Monomeric RAS: HRAS Costello syndrome KRAS Noonan syndrome 3 KRAS Cardiofaciocutaneous syndrome RAB: RAB7 Charcot–Marie–Tooth disease RAB23 Carpenter syndrome RAB27 Griscelli syndrome type 2 RHO: RAC2 Neutrophil immunodeficiency syndrome ARF : SAR1B Chylomicron retention disease ARL13B Joubert syndrome 8 ARL6 Bardet–Biedl syndrome 3 MAP kinase Cardiofaciocutaneous syndrome Other kinase / phosphatase Tyrosine kinase BTK X-linked agammaglobulinemia ZAP70 ZAP70 deficiency Serine/threonine kinase RPS6KA3 Coffin-Lowry syndrome CHEK2 Li-Fraumeni syndrome 2 IKBKG Incontinentia pigmenti STK11 Peutz–Jeghers syndrome DMPK Myotonic dystrophy 1 ATR Seckel syndrome 1 GRK1 Oguchi disease 2 WNK4 / WNK1 Pseudohypoaldosteronism 2 Tyrosine phosphatase PTEN Bannayan–Riley–Ruvalcaba syndrome Lhermitte–Duclos disease Cowden syndrome Proteus-like syndrome MTM1 X-linked myotubular myopathy PTPN11 Noonan syndrome 1 LEOPARD syndrome Metachondromatosis Signal transducing adaptor proteins EDARADD EDARADD Hypohidrotic ectodermal dysplasia SH3BP2 Cherubism LDB3 Zaspopathy Other NF2 Neurofibromatosis type II NOTCH3 CADASIL PRKAR1A Carney complex PRKAG2 Wolff–Parkinson–White syndrome PRKCSH PRKCSH Polycystic liver disease XIAP XIAP2 See also intracellular signaling peptides and proteinsNOTCH3, HTRA1, EGF, COL4A1, COL18A1, NOTCH1, JAG1, APOE, APP, LPA, TREX1, GFAP, PLXNA2, CTSA, PROC, ACTB, SOD1, TGFB1, NOTCH2, LAP, MRS2, RNF213, NOX5, THBD, KDR, NEFL, ACTG2, RBPJ, FN1, FBN1, ELN, DVL1, DCN, CSF3, CSF1R, CLU, CACNA1A, BGN, APCS, LINC01191