Ankyrin-B syndrome is associated with a variety of heart problems related to disruption of the heart's normal rhythm (arrhythmia). ... In people with ankyrin-B syndrome, arrhythmia can lead to fainting (syncope) or cardiac arrest and sudden death. When associated with a prolonged QT interval, the condition is sometimes classified as long QT syndrome 4. However, because additional heart problems can result from changes in the same gene, long QT syndrome 4 is usually considered part of ankyrin-B syndrome. Frequency Ankyrin-B syndrome is a rare disorder. Its prevalence is unknown. ... Learn more about the gene associated with Ankyrin-B syndrome ANK2 Inheritance Pattern Ankyrin-B syndrome is inherited in an autosomal dominant pattern , which means one copy of the altered ANK2 gene in each cell is sufficient to cause the disorder.
Isaac's syndrome is an immune-mediated peripheral motor neuron disorder characterized by continuous muscle fiber activity at rest resulting in muscle stiffness, cramps, myokymia, and pseudomyotonia. ... Diagnostic methods Diagnosis of Isaac's syndrome relies on history, physical findings, and electromyography (EMG). Typical findings are doublet, triplet or multiplet ('myokymic') motor unit discharges and fasciculations. 35-40% of patients have elevated levels of anti-VGKC-complex antibodies, which is more frequent in Morvan syndrome than in Isaac's syndrome. Differential diagnosis Differential diagnosis includes hereditary neuromyotonia or myokymia (with/without episodic ataxia) syndromes, cramp-fasciculation syndrome, motor neuron diseases (progressive spinal muscle atrophy, neuropathy, amyotrophic lateral sclerosis (ALS) (see this term), intoxication (gold, mercury, toluene, insecticides) and tetanus (see this term). ... Prognosis There is no cure for Isaac's syndrome although it is not fatal. Morvan's syndrome has a more severe course and can be fatal. The long-term prognosis for individuals with paraneoplastic Isaac's syndrome is dependent on the tumor course.
History Gamstorp and Wohlfart (1959) described a syndrome of myokymia (fine muscle twitches), myotonia, muscle wasting, and hyperhidrosis in 3 unrelated patients.
Another feature of this condition is neuromyotonia (also known as Isaac syndrome). Neuromyotonia results from overactivation (hyperexcitability) of peripheral nerves, which leads to delayed relaxation of muscles after voluntary tensing (contraction), muscle cramps, and involuntary rippling movement of the muscles (myokymia).
Isaacs' syndrome is a rare neuromuscular disorder that is characterized by progressive muscle stiffness; continuously contracting or twitching muscles (myokymia); and diminished reflexes.
A rare peripheral neuropathy characterized by slowly progressive axonal, motor greater than sensory, polyneuropathy combined with neuromytonia (including spontaneous muscular activity at rest (myokymia), impaired muscle relaxation (pseudomyotonia), and contractures of hands and feet) and neuromyotonic or myokymic discharges on needle EMG. It presents with distal lower limb weakness with gait impairment, muscle stiffness, fasciculations and cramps in hands and legs worsened by cold, decreased to absent tendon reflexes, intrinsic hand muscle atrophy and, variably, mild distal sensory impairment.
Swyer syndrome is a condition that affects sexual development. ... People with Swyer syndrome have typical female external genitalia. ... Women with this disorder do not produce eggs (ova), but they may be able to become pregnant with a donated egg or embryo. Swyer syndrome usually affects only sexual development; such cases are called isolated Swyer syndrome. ... Frequency Swyer syndrome occurs in approximately 1 in 80,000 people. ... However, in most individuals with Swyer syndrome, the cause is unknown. Learn more about the genes associated with Swyer syndrome DHH MAP3K1 NR0B1 NR5A1 SOX9 SRY Additional Information from NCBI Gene: CBX2 DMRT1 ZFPM2 Inheritance Pattern Most cases of Swyer syndrome are not inherited; they occur in people with no history of the condition in their family.
Primary empty sella syndrome occurs when a small anatomical defect above the pituitary gland increases pressure in the sella turcica and causes the gland to flatten out along the interior walls of the sella turcica cavity. [3] Primary empty sella syndrome is associated with obesity and increase in intracranial pressure in women. [9] In most cases, especially in people with primary empty sella syndrome, there are no symptoms and it does not affect life expectancy or health. ... Retrieved 6 March 2017 . ^ a b c "Empty Sella Syndrome Information Page" . www.ninds.nih.gov . ... Retrieved 2017-03-05 . ^ a b c "Empty sella syndrome" . rarediseases.info.nih.gov . ... ISBN 9780826198549 . ^ Fouad, Wael (2011-06-01). "Review of empty sella syndrome and its surgical management" . ... External links [ edit ] Classification D ICD - 10 : E23.6 ICD - 9-CM : 253.8 MeSH : D004652 DiseasesDB : 31523 SNOMED CT : 237722004 External resources MedlinePlus : 000349 Orphanet : 91354 Scholia has a topic profile for Empty sella syndrome . v t e Pituitary disease Hyperpituitarism Anterior Acromegaly Hyperprolactinaemia Pituitary ACTH hypersecretion Posterior SIADH General Nelson's syndrome Hypophysitis Hypopituitarism Anterior Kallmann syndrome Growth hormone deficiency Hypoprolactinemia ACTH deficiency / Secondary adrenal insufficiency GnRH insensitivity FSH insensitivity LH/hCG insensitivity Posterior Neurogenic diabetes insipidus General Empty sella syndrome Pituitary apoplexy Sheehan's syndrome Lymphocytic hypophysitis Pituitary adenoma
Empty sella syndrome (ESS) is a condition that involves the sella turcica, a bony structure at the base of the brain that protects the pituitary gland .
Summary Clinical characteristics. Spinocerebellar ataxia type 13 (SCA13) is a phenotypic spectrum that includes both non-progressive infantile-onset ataxia and progressive childhood-onset and adult-onset cerebellar ataxia. Three phenotypes are seen: Cerebellar hypoplasia with non-progressive infantile-onset limb, truncal, and gait ataxia with mild-to-moderate intellectual disability and occasionally seizures and/or psychiatric manifestations. Cognition and motor skills improve over time. Childhood-onset slowly progressive cerebellar atrophy with slowly progressive cerebellar ataxia and dysarthria, delayed motor milestones, and mild-to-moderate intellectual disability Adult-onset progressive cerebellar atrophy with progressive ataxia and spasticity Diagnosis/testing. The diagnosis of spinocerebellar ataxia type 13 (SCA13) is established in a proband with suggestive clinical and brain imaging findings and a heterozygous KCNC3 pathogenic variant identified by molecular genetic testing. Management. Treatment of manifestations: A multidisciplinary approach to management of ataxia and related neurologic manifestations is recommended including neurology, physical therapy (PT), occupational therapy (OT), speech and language pathology, and feeding team, as well as experts in educational needs and/or social/behavioral issues.
A number sign (#) is used with this entry because of evidence that spinocerebellar ataxia-13 (SCA13) is caused by heterozygous mutation in the KCNC3 gene (176264) on chromosome 19q13. For a general discussion of autosomal dominant spinocerebellar ataxia, see SCA1 (164400). Clinical Features Herman-Bert et al. (2000) described a large French family in which 8 members were affected with autosomal dominant spinocerebellar ataxia. There were 7 affected women and an affected 4-year-old boy. Clinical features included slowly progressive childhood-onset cerebellar gait ataxia associated with cerebellar dysarthria, moderate mental retardation (IQ, 62-76), and mild developmental delay in motor acquisition. Nystagmus and pyramidal signs were also observed in some patients. Cerebral magnetic resonance imaging in 2 patients showed moderate cerebellar and pontine atrophy.
Spinocerebellar ataxia 13 (SCA13) is a rare sub-type of spinocerebellar ataxias (SCA), a group of neurological diseases that causes degeneration of the brain and spinal cord. The age when symptoms begin and the type and severity of symptoms of SCA13 can be different from person to person even in the same family. In almost every case, the disease progresses very slowly and does not affect a person's life span. Symptoms most commonly begin in early childhood or later in midlife, but can range from infancy to 60. The childhood forms is often least progressive, but may also include mild to moderate learning problems, taking longer to learn to crawl, walk, or run (delayed development of motor skills), and seizures.
Clinical description SCA13 is primarily a cerebellar syndrome, but dysphagia, urinary urgency, and bradykinesia have been described in affected patients older than 50.
Wieacker syndrome Other names Intellectual disability-developmental delay-contractures syndrome This condition is inherited in an X-linked recessive manner. Wieacker Syndrome or Wieacker-Wolff syndrome is a rare, severely disabling, genetic disorder. ... Genetics [ edit ] Wieacker syndrome is caused by a mutation in ZC4H2 on the X chromosome (Xq13-q21). [2] Diagnosis [ edit ] This section is empty. ... There are no effective disease-modifying therapies. [2] Epidemiology [ edit ] Fewer than 30 cases have been identified. [3] References [ edit ] ^ "OMIM Entry # 314580 - WIEACKER-WOLFF SYNDROME; WRWF" . www.omim.org . ^ a b c "Wieacker Syndrome - NORD (National Organization for Rare Disorders)" . ^ RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Intellectual disability developmental delay contractures syndrome" . www.orpha.net . External links [ edit ] Classification D OMIM : 314580 MeSH : C536703 External resources Orphanet : 3454
Intellectual disability-developmental delay-contractures syndrome is a rare, slowly progressive genetic disorder that is present at birth. ... Intellectual disability-developmental delay-contractures syndrome is caused by mutations in the ZC4H2 gene and is inherited in an X-linked recessive fashion. Most people with intellectual disability-developmental delay-contractures syndrome are male; however carrier females have been reported to have mild symptoms. There is no known cure for intellectual disability-developmental delay-contractures syndrome. Treatment is symptomatic and supportive.
Intellectual disability-developmental delay-contractures syndrome, formerly known as Wieacker-Wolff syndrome, is a severe X-linked recessive neurodevelopmental disorder characterized by severe contractures (arthrogryposis; see this term) and intellectual disability. Epidemiology Prevalence and incidence rates are not known. The syndrome has been reported in 5 families to date, with fewer than 30 affected individuals described. ... Cases have been reported from Germany, France, the Netherlands, Australia, and the United States. Clinical description The syndrome is characterized by an association of arthrogryposis multiplexa congenita and intellectual disability. ... Etiology Intellectual disability-developmental delay-contractures syndrome is caused by mutations in the ZC4H2 gene (Xq11.1) that is presumed to play a role in neuronal function during fetal growth.
Severe insulin resistance underlies the varied signs and symptoms of Donohue syndrome. Individuals with Donohue syndrome are unusually small starting before birth, and affected infants experience failure to thrive, which means they do not grow and gain weight at the expected rate. ... Affected individuals develop recurrent, life-threatening infections beginning in infancy. Donohue syndrome is one of a group of related conditions described as inherited severe insulin resistance syndromes. These disorders, which also include Rabson-Mendenhall syndrome and type A insulin resistance syndrome, are considered part of a spectrum. Donohue syndrome represents the most severe end of the spectrum; most children with this condition do not survive beyond age 2. Frequency Donohue syndrome is estimated to affect less than 1 per million people worldwide.
The signs and symptoms of Crouzon syndrome with acanthosis nigricans overlap with those of a similar condition called Crouzon syndrome. ... Less common features that can occur in either disorder include an opening in the roof of the mouth (cleft palate ), dental problems, or hearing loss. People with Crouzon syndrome or Crouzon syndrome with acanthosis nigricans usually have normal intelligence. Crouzon syndrome with acanthosis nigricans is distinguished from Crouzon syndrome by several features, including skin abnormalities. ... Nasal passage abnormalities and hydrocephalus are rare in Crouzon syndrome. Less common features of Crouzon syndrome with acanthosis nigricans include subtle changes in the bones of the spine (vertebrae), abnormalities of the finger bones, and noncancerous growths in the jaw called cementomas. Frequency Crouzon syndrome with acanthosis nigricans is rare; this condition occurs in about 1 person per million.
Crouzon syndrome with acanthosis nigricans (CAN) is a very rare, clinically heterogeneous form of faciocraniostenosis with Crouzon-like features and premature synostosis of cranial sutures (Crouzon disease, see this term), associated with acanthosis nigricans (AN; see this term). ... Clinical description All patients have congenital craniofacial abnormalities consistent with classic Crouzon syndrome including craniosynostosis, midface hypoplasia, shallow orbits with exophthalmos, down-slanting palpebral fissures and hypertelorism, maxillary hypoplasia with convex nose and posteriorly angulated ears. ... Some of these specific features are rare in patients with classic Crouzon syndrome. Intellectual disability, hearing loss and speech delay are uncommon.
Crouzonodermoskeletal syndrome Other names Crouzon syndrome with acanthosis nigricans Crouzonodermoskeletal syndrome is a disorder characterized by the premature joining of certain bones of the skull ( craniosynostosis ) during development and a skin condition called acanthosis nigricans . [1] Some of the signs and symptoms of Crouzonodermoskeletal syndrome are similar to those seen with Crouzon syndrome . ... People with these conditions are generally of normal intelligence. [2] Several features distinguish Crouzonodermoskeletal syndrome from Crouzon syndrome. People with Crouzonodermoskeletal syndrome have acanthosis nigricans, a skin condition characterized by thick, dark, velvety skin in body folds and creases, including the neck and underarms. ... You can help by adding to it . ( October 2017 ) References [ edit ] ^ Herman, TE; Sargar, K; Siegel, MJ (Feb 2014). "Crouzono-dermo-skeletal syndrome, Crouzon syndrome with acanthosis nigricans syndrome". ... "Subtle radiographic findings of achondroplasia in patients with Crouzon syndrome with acanthosis nigricans due to an Ala391Glu substitution in FGFR3". ... "Let's call it "Crouzonodermoskeletal syndrome" so we won't be prisoners of our own conventional terminology".
Description Crouzon syndrome with acanthosis nigricans is considered to be a distinct disorder from classic Crouzon syndrome (123500), which is caused by mutation in the FGFR2 gene (176943). Cohen (1999) argued that this condition is separate from Crouzon syndrome for 2 main reasons: it is caused by a highly specific mutation of the FGFR3 gene, whereas multiple different FGFR2 mutations result in Crouzon syndrome, and the phenotypes are different. ... Schweitzer et al. (2001) concluded that the presence of choanal atresia and hydrocephalus in an individual with features of Crouzon syndrome should suggest the diagnosis of Crouzon syndrome with acanthosis nigricans, even before the skin involvement appears. ... This mutation was not present in 16 unrelated Crouzon syndrome patients with FGFR2 mutations, 13 unrelated Crouzon syndrome patients without FGFR2 IgIII domain mutations, or 50 unrelated controls. ... In 1 kindred, a patient with Crouzon syndrome with acanthosis nigricans had a second cousin with classic Crouzon syndrome due to an FGFR2 mutation (S347C; 176943.0009), and the authors raised the possibility of a predisposing factor for FGFR mutations.
See also: Autoimmune polyendocrine syndrome type 1 Autoimmune polyendocrine syndrome type 2 Other names Schmidt's syndrome [1] HLA-DQ2 one of the human leukocyte antigens genotypes responsible for this condition Specialty Endocrinology Symptoms Asplenia [1] Risk factors Human leukocyte antigen (HLA-DQ2, HLA-DQ8 and HLA-DR4) [2] Diagnostic method Ultrasound, MRI [3] Treatment Thyroid-stimulating hormone [4] Autoimmune polyendocrine syndrome type 2 , a form of autoimmune polyendocrine syndrome also known as APS-II, or PAS II, is the most common form of the polyglandular failure syndromes. [2] PAS II is defined as the association between autoimmune Addison's disease and either autoimmune thyroid disease , type 1 diabetes , or both. [5] It is heterogeneous and has not been linked to one gene . ... Because of this, both of these subtypes are generally referred to as PAS II. [13] [14] See also [ edit ] IPEX syndrome Autoimmune polyendocrine syndrome References [ edit ] ^ a b c d "Autoimmune polyglandular syndrome type 2 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ... "Update on autoimmune polyendocrine syndromes (APS)". Acta Biomed . 74 (1): 9–33. ... "Autoimmune polyglandular syndrome, type II" . American Family Physician . 75 (5): 667–70. ... S. (2009-05-01). "Autoimmune polyendocrine syndrome type 1 (APS-1) as a model for understanding autoimmune polyendocrine syndrome type 2 (APS-2)" .
Description Autoimmune polyendocrine syndrome type II (APS2), or Schmidt syndrome, is characterized by the presence of autoimmune Addison disease in association with either autoimmune thyroid disease or type I diabetes mellitus, or both. ... See 240300 for a phenotypic description of autoimmune polyendocrine syndrome type I (APS1). Clinical Features Carpenter et al. (1964) reviewed the literature on Schmidt syndrome and reported 15 new cases. ... Eisenbarth and Gottlieb (2004) compared the features of 3 autoimmune polyendocrine syndromes: autoimmune polyendocrine syndrome type I (240300), autoimmune polyendocrine syndrome type II, or Schmidt syndrome, and X-linked polyendocrinopathy with immune dysfunction and diarrhea (304790). Mapping Eisenbarth et al. (1978) found evidence of an association of HLA-B8 (see HLA-B, 142830) with the polyglandular failure syndrome in 3 generations of a family. The 10 unaffected individuals did not have B8, and only 1 of 7 members with B8 escaped the syndrome. ... Patients with type I autoimmune polyglandular syndrome did not show the association. In the majority of cases, Addison disease is a component of an autoimmune polyendocrine syndrome, or APS (Gambelunghe et al., 1999).
A rare, endocrine disease characterized by autoimmune Addison disease associated with autoimmune thyroid disease or type I diabetes mellitus, or both, and without chronic candidiasis. Additional endocrine (hypogonadism, hypoparathyroidism) and non-endocrine diseases (vitiligo, autoimmune hepatitis, autoimmune gastritis, pernicious anemia, and myasthenia gravies) may be present.
Autoimmune polyglandular syndrome type 2 is an autoimmune disorder that affects many hormone-producing ( endocrine ) glands. ... Affected individuals may also have problems with other endocrine glands and other common features include primary hypogonadism , myasthenia gravis , and celiac disease . Autoimmune polyglandular syndrome type 2 is diagnosed in adulthood, typically around age 30. The cause of autoimmune polyglandular syndrome type 2 is unknown, although it may involve a combination of genetic and environmental factors.
SRTD encompasses Ellis-van Creveld syndrome (EVC) and the disorders previously designated as Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib-polydactyly syndrome (SRPS), and Mainzer-Saldino syndrome (MZSDS). ... There is phenotypic overlap with the cranioectodermal dysplasias (Sensenbrenner syndrome; see CED1, 218330). For a discussion of genetic heterogeneity of short-rib thoracic dysplasia, see SRTD1 (208500). ... Due to the combination of thoracic hypoplasia with pelvic and phalangeal abnormalities, a clinical diagnosis of Jeune syndrome was made. Halbritter et al. (2013) studied 14 patients from 12 families with mutations in the IFT172 gene who shared a phenotype that included skeletal abnormalities, nephronophthisis, and liver and eye involvement, consistent with a clinical diagnosis of complex asphyxiating thoracic dystrophy or Mainzer-Saldino syndrome. ... Both Filipino sibs and a female Hungarian patient also had cerebellar vermis hypoplasia, which Halbritter et al. (2013) noted was a classic feature of Joubert syndrome (see 213300). In addition, 3 of the patients with an MZSDS phenotype as well as the female Hungarian patient exhibited obesity and impaired glucose tolerance, suggesting phenotypic overlap with Bardet-Biedl syndrome (209900). ... Sanger sequencing of exons and intron-exon boundaries of the 14 IFTB-complex genes in another cohort of 296 individuals with ciliopathies revealed compound heterozygous mutations in IFT172 (607386.0001; 607386.0008) in a Belgian girl with a clinical diagnosis of Jeune syndrome, originally described by Casteels et al. (2000).
Biallelic mutations in the CEP120 gene have also been reported in patients with Joubert syndrome (JBTS31; 617761). For a general phenotypic description and discussion of genetic heterogeneity of short-rib thoracic dysplasia with or without polydactyly, see SRTD1 (208500). ... Roosing et al. (2016) reported a male fetus of Flemish origin (SW-476410) diagnosed in utero with tectal and cerebellar dysraphia with occipital encephalocele (TCDOE) that was considered to be 'in the spectrum of Joubert syndrome' due to the presence of a suboccipital encephalocele, dysplastic tectum, severe hypoplasia of the cerebellar vermis, and the molar tooth sign seen on fetal MRI at 23 weeks' gestation. ... Roosing et al. (2016) also studied a male fetus of Turkish origin (MKS-2930) initially diagnosed with Meckel syndrome (see 249000) in the second trimester of pregnancy, due to the presence of cystic dysplastic kidneys, occipital encephalocele, and polydactyly. ... By exome sequencing in another fetus of Turkish origin (MKS-2930), initially diagnosed with Meckel syndrome but also showing features of SRTD with polydactyly, Roosing et al. (2016) identified homozygosity for a missense mutation in CEP120 (I949S; 613446.0003). Contrasting the severity of the phenotype in these patients with the relatively mild, purely neurologic phenotype in 4 patients with CEP120-associated Joubert syndrome (JBTS31; 617761), Roosing et al. (2016) stated that the mechanism through which mutations in the same gene cause such wide phenotypic variability remained unexplained.
SRTD encompasses Ellis-van Creveld syndrome (EVC) and the disorders previously designated as Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib-polydactyly syndrome (SRPS), and Mainzer-Saldino syndrome (MZSDS). ... There is phenotypic overlap with the cranioectodermal dysplasias (Sensenbrenner syndrome; see CED1, 218330). For a discussion of genetic heterogeneity of short-rib thoracic dysplasia, see SRTD1 (208500). Clinical Features De Vries et al. (2010) described 13 patients with Jeune syndrome, including a 22-year-old Dutch woman who was born to healthy, unrelated parents and had 1 healthy younger sib. ... Molecular Genetics Bredrup et al. (2011) analyzed the candidate gene WDR19 (608151) in 14 patients with Jeune syndrome. In a 22-year-old Dutch woman originally reported by de Vries et al. (2010), they identified homozygosity for a missense mutation in the WDR19 gene (L7P; 608151.0003).
SRTD encompasses Ellis-van Creveld syndrome (EVC) and the disorders previously designated as Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib-polydactyly syndrome (SRPS), and Mainzer-Saldino syndrome (MZSDS). ... There is phenotypic overlap with the cranioectodermal dysplasias (Sensenbrenner syndrome; see CED1, 218330). For a discussion of genetic heterogeneity of short-rib thoracic dysplasia, see SRTD1 (208500). ... In a patient with a clinical diagnosis of lethal short-rib polydactyly syndrome, Cavalcanti et al. (2011) identified a homozygous missense mutation in the IFT80 gene (611177.0004).
SRTD encompasses Ellis-van Creveld syndrome (EVC) and the disorders previously designated as Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib-polydactyly syndrome (SRPS), and Mainzer-Saldino syndrome (MZSDS). ... Labrune et al. (1999) reported 3 children with Jeune syndrome who had clinical and laboratory evidence of liver disease. ... Singh et al. (1988) described 4 patients, including 2 sibs, with Jeune syndrome and mild congenital hydrocephalus. ... Rinaldi et al. (1990) reported 2 sisters who had both Jeune syndrome and cystinuria (220100). The parents, living in Italy, were presumably unrelated. ... Tuysuz et al. (2009) reported 10 patients with Jeune syndrome, all of whom were born of consanguineous parents.
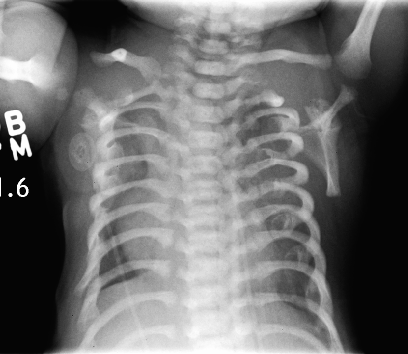
Jeune syndrome is a rare condition that primarily affects the bones. ... In many cases the cause of Jeune syndrome is unknown; however, changes (mutations) in several different genes ( IFT80 , DYNC2H1 , WDR19 , IFT140 and TTC21B ) have been identified in some families with the condition. Jeune syndrome is inherited in an autosomal recessive manner.
SRTD encompasses Ellis-van Creveld syndrome (EVC) and the disorders previously designated as Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib-polydactyly syndrome (SRPS), and Mainzer-Saldino syndrome (MZSDS). ... There is phenotypic overlap with the cranioectodermal dysplasias (Sensenbrenner syndrome; see CED1, 218330). For a discussion of genetic heterogeneity of short-rib thoracic dysplasia, see SRTD1 (208500).
Jeune syndrome, also called asphyxiating thoracic dystrophy, is a short-rib dysplasia characterized by a narrow thorax, short limbs and radiological skeletal abnormalities including 'trident' aspect of the acetabula and metaphyseal changes. Epidemiology Annual incidence at birth is unknown but is estimated to be 1-5/500,000. Clinical description The syndrome is recognizable during the antenatal period or at birth. ... Etiology The molecular basis of the syndrome has been partially elucidated indicating involvement of the IFT80 (3q25.33), DYNC2H1 (11q22.3), WDR19 (4p14) and TTC21B (2q24.3) genes, each encoding an intraflagellar transport protein, which confirms that Jeune syndrome belongs to the ciliopathies group. ... Differential diagnosis Differential diagnosis should include thoracolaryngopelvic dysplasia, Ellis-van Creveld syndrome, Sensenbrenner syndrome and paternal uniparental disomy of chromosome 14 (see these terms). ... In other cases, only a careful antenatal ultrasound examination can detect the disease. Genetic counseling The syndrome is transmitted as an autosomal recessive trait.
Familial atrial fibrillation is a rare, genetically heterogenous cardiac disease characterized by erratic activation of the atria with an irregular ventricular response, in various members of a single family. It may be asymptomatic or associated with palpitations, dyspnea and light-headedness. Concomitant rhythm disorders and cardiomyopathies are frequently reported.
SCARF syndrome Other names Skeletal abnormalities, Cutis laxa, craniostenosis, Ambiguous genitalia, Retardation, and Facial abnormalities [1] This condition is inherited in an X-linked recessive manner SCARF syndrome is a rare syndrome characterized by skeletal abnormalities, cutis laxa , craniostenosis , ambiguous genitalia , psychomotor retardation , and facial abnormalities. ... The relationship of the parents as well as family history of X-linked , autosomal dominant , and autosomal disorders should be considered to confirm SCARF syndrome. [3] Treatment/Prevention [ edit ] Due to the fact that the syndrome is a genetic condition, there is no specific cure for this syndrome. ... References [ edit ] ^ "SCARF syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . Retrieved 28 June 2019 . ^ "Scarf Syndrome disease: Malacards - Research Articles, Drugs, Genes, Clinical Trials" . www.malacards.org . ... Retrieved 2020-11-12 . ^ a b "Orphanet: SCARF syndrome" . www.orpha.net . Retrieved 2020-11-12 . ^ Mathijssen, Irene.
A rare multiple congenital anomalies syndrome characterized by variable skeletal abnormalities (including craniostenosis, pectus carinatum, short sternum, joint hyperextensibility, and anbnormal vertebrae), cutis laxa with excessive skin folds around the cheek, chin and neck, ambiguous genitalia with a micropenis and perineal hypospadia, an umbilical hernia, intellectual disability, premature aged appearance, and cardiac enlargement involving either the ventricles or atria.
Ulnar tunnel syndrome Other names Guyon's canal syndrome Cartoon depiction of classic ulnar sensory distribution, including mid-4th and 5th fingers. ... Specialty Orthopedic surgery Ulnar tunnel syndrome , also known as Guyon's canal syndrome or Handlebar palsy , is caused by entrapment of the ulnar nerve in the Guyon canal as it passes through the wrist. [1] Symptoms usually begin with a feeling of pins and needles in the ring and little fingers before progressing to a loss of sensation and/or impaired motor function of the intrinsic muscles of the hand which are innervated by the ulnar nerve . Ulnar tunnel syndrome is commonly seen in regular cyclists due to prolonged pressure of the Guyon's canal against bicycle handlebars. ... "Syndrome of canal of Guyon - definition, diagnosis, treatment and complication". ... US patent 6845514 , Yao, Joseph, "Protective device for the median and ulnar nerves", issued January 25, 2005 v t e Diseases relating to the peripheral nervous system Mononeuropathy Arm median nerve Carpal tunnel syndrome Ape hand deformity ulnar nerve Ulnar nerve entrapment Froment's sign Ulnar tunnel syndrome Ulnar claw radial nerve Radial neuropathy Wrist drop Cheiralgia paresthetica long thoracic nerve Winged scapula Backpack palsy Leg lateral cutaneous nerve of thigh Meralgia paraesthetica tibial nerve Tarsal tunnel syndrome plantar nerve Morton's neuroma superior gluteal nerve Trendelenburg's sign sciatic nerve Piriformis syndrome Cranial nerves See Template:Cranial nerve disease Polyneuropathy and Polyradiculoneuropathy HMSN Charcot–Marie–Tooth disease Dejerine–Sottas disease Refsum's disease Hereditary spastic paraplegia Hereditary neuropathy with liability to pressure palsy Familial amyloid neuropathy Autoimmune and demyelinating disease Guillain–Barré syndrome Chronic inflammatory demyelinating polyneuropathy Radiculopathy and plexopathy Brachial plexus injury Thoracic outlet syndrome Phantom limb Other Alcoholic polyneuropathy Other General Complex regional pain syndrome Mononeuritis multiplex Peripheral neuropathy Neuralgia Nerve compression syndrome
They designated the disorder 'fertile eunuch' syndrome. One patient had a brother with eunuchoidal features who refused examination. ... Pasqualini (1953) reported a 24-year-old Argentinian man who had a well-defined 'hypoandrogenic' syndrome. He exhibited lipomastia without gynecomastia, lacked facial and body hair, and had scant pubic and axillary hair. ... INHERITANCE - Autosomal recessive HEAD & NECK Nose - Normal sense of smell CHEST Breasts - Gynecomastia GENITOURINARY External Genitalia (Male) - Small penis Internal Genitalia (Male) - Small or normal testes - Low semen volume - Reduced or absent spermatogenesis - Reduced or absent Leydig cells - Testicular microlithiasis Internal Genitalia (Female) - Oligomenorrhea after menarche - Secondary amenorrhea - Small to normal uterus - Multicystic ovaries SKIN, NAILS, & HAIR Hair - Sparse to absent male facial hair - Sparse to absent axillary hair - Sparse to absent pubic hair VOICE - Juvenile voice ENDOCRINE FEATURES - Hypogonadotropic hypogonadism - Delayed or absent puberty in males - Spontaneous normal puberty in females - Low to undetectable luteinizing hormone levels - Normal to high follicle-stimulating hormone (FSH) levels - Inappropriately normal inhibin-B (INHBB) levels - Low testosterone levels - Low to normal estradiol levels MISCELLANEOUS - Some male patients exhibit some degree of spermatogenesis, hence the designation 'fertile eunuch syndrome' has been used - Affected females have apparently normal puberty but later develop secondary amenorrhea with anovulatory cycles MOLECULAR BASIS - Caused by mutation in the luteinizing hormone beta-polypeptide gene (LHB, 152780.0001 ) ▲ Close
Secondary Evans syndrome was associated with higher mortality rate than primary Evans syndrome, with a 5-year survival of 38%. Among patients with Evans syndrome, the prevailing causes of death were bleeding, infections, and hematological cancer. [4] It has been observed that there is a risk of developing other autoimmune problems and hypogammaglobulinemia , [28] in one cohort 58% of children with Evans syndrome had CD4-/CD8- T cells which is a strong predictor for having autoimmune lymphoproliferative syndrome . [29] Epidemiology [ edit ] Evans syndrome is considered a very rare autoimmune disease. ... PMID 1451398 . ^ "EVANS SYNDROME" . 2019-10-22. Cite journal requires |journal= ( help ) ^ "Evans syndrome" . ... PMID 16124452 . ^ Mathew P, Chen G, Wang W (1997). "Evans syndrome: results of a national survey". ... S2CID 20125761 . ^ Teachey DT; Manno CS; Axsom KM; et al. (2005). "Unmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS)" .
Evans syndrome is a very rare autoimmune disorder in which the immune system destroys the body's red blood cells, white blood cells and/or platelets. ... The best treatment options for Evans syndrome depend on many factors, including the severity of the condition; the signs and symptoms present; and each person's response to certain therapies.
Epidemiology The prevalence in Europe is estimated at 1/1,000,000 but there are no robust epidemiological data available. Clinical description The syndrome can manifest both in childhood or adulthood. ... AIHA manifests as an unusual weakness, pallor, fatigue with tachycardia and exertional dyspnea, and also in some cases jaundice, dark urine and/or splenomegaly. Etiology Evans syndrome is an autoimmune disorder in which non-cross-reacting autoantibodies are targeted towards different antigenic determinants on red cells, platelets, sometimes neutrophils; however, the exact pathophysiologic mechanism is unknown. Because of the observation of a decrease in T-helper and an increase in T-suppressor lymphocyte population, it is suggested that the cytopenia may be related to T-cell abnormalities. Evans syndrome is frequently associated with other diseases, such as systemic lupus erythematosus, antiphospholipid syndrome, autoimmune lymphoproliferative syndrome, and common variable immunodeficiency (see these terms), which could point to a common cellular and humoral abnormality. ... Differential diagnosis Differential diagnosis mainly includes microangiopathies such as thrombotic thrombocytopenic purpura, and typical or atypical hemolytic uremic syndrome (see these terms). Genetic counseling Most of the cases are sporadic. ... In severe cases, hematopoietic stem cell transplantation may be required. Prognosis Evans syndrome is a chronic disease with alternating periods of remission and relapse of AIHA and/or ITP despite treatment, which can be associated with significant morbidity and mortality due to severe hemorrhage and infections in case of severe thrombocytopenia and neutropenia.
Not to be confused with McLeod syndrome . Swyer–James syndrome Other names Macleod syndrome, Swyer–James–Macleod's syndrome Swyer–James syndrome of the left (smaller) lung, virtual CT-bronchography. Specialty Respirology Swyer–James syndrome ( SJS , also called MacLeod syndrome ) is a rare lung disorder found by English chest physician William Mathiseon Macleod, [1] and (simultaneously) by physician Paul Robert Swyer and radiologist George James in the 1950s in Canada. [2] Swyer–James syndrome is a manifestation of postinfectious obliterative bronchiolitis . ... PMID 13077508 . ^ Braunschweig M, Gal I (April 2001). "Swyer-James syndrome". JBR-BTR . 84 (2): 57. PMID 11374630 . ^ Chalmers JH (September 1999). "Swyer-James syndrome". Seminars in Respiratory Infections . 14 (3): 295–7. ... PMID 18460255 . ^ Gopinath A, Strigun D, Banyopadhyay T (2005). "Swyer-James syndrome". Connecticut Medicine . 69 (6): 325–7.
Swyer-James syndrome is a rare condition in which the lung (or portion of the lung) does not grow normally and is slightly smaller than the opposite lung, usually following bronchiolitis in childhood.
The disorder is inherited in an autosomal recessive manner. This syndrome is divided into types I and II, with the latter sometimes called Arias syndrome. These two types, along with Gilbert's syndrome , Dubin–Johnson syndrome , and Rotor syndrome , make up the five known hereditary defects in bilirubin metabolism. Unlike Gilbert's syndrome, only a few causes of Crigler-Najjar syndrome are known. ... PMID 10603107 . ^ Crigler Najjar Syndrome. National Organization for Rare Disorders (NORD) . 2016; https://rarediseases.org/rare-diseases/crigler-najjar-syndrome/. ^ "AT342 – Crigler-Najjar Syndrome – Audentes Therapeutics" . www.audentestx.com . ... "Treatment of the Crigler–Najjar syndrome type I with hepatocyte transplantation".
Crigler-Najjar syndrome is a severe condition characterized by high levels of a toxic substance called bilirubin in the blood (hyperbilirubinemia). ... Bilirubin has an orange-yellow tint, and hyperbilirubinemia causes yellowing of the skin and whites of the eyes (jaundice). In Crigler-Najjar syndrome, jaundice is apparent at birth or in infancy. ... Mutations in the UGT1A1 gene that cause Crigler-Najjar syndrome result in reduced or absent function of the bilirubin-UGT enzyme. ... Learn more about the gene associated with Crigler-Najjar syndrome UGT1A1 Inheritance Pattern Crigler-Najjar syndrome is inherited in an autosomal recessive pattern , which means both copies of the UGT1A1 gene in each cell have mutations. A less severe condition called Gilbert syndrome can occur when one copy of the UGT1A1 gene has a mutation.
Mutations in the same gene cause Gilbert syndrome (143500) and Crigler-Najjar syndrome type II (606785). ... In Crigler-Najjar syndrome, intense jaundice appears in the first days of life and persists thereafter. ... The level of total serum bilirubin, predominantly indirect-reacting, ranges from 20 to 45 mg/dL. In Crigler-Najjar syndrome type II, total serum bilirubin ranges from 6 to 20 mg/dL, and in Gilbert syndrome, total serum bilirubin ranges from 1 to 6 mg/dL. ... A review of glucuronidation research in relation to the Crigler-Najjar syndrome was provided by Jansen et al. (1992). ... In 2 offspring, a boy and a girl, of first-cousin Saudi parents, Nazer et al. (1990) described the concurrence of Robinow syndrome and Crigler-Najjar syndrome. Two previously born children had died at the age of 2 months with progressive jaundice but without the fetal facial characteristics of Robinow syndrome.
Crigler Najjar syndrome, type 1 is an inherited disorder in which bilirubin , a substance made by the liver, cannot be broken down. ... Without this enzyme, bilirubin can build up in the body and lead to jaundice and damage to the brain, muscles, and nerves. Crigler Najjar syndrome, type 1 is caused by mutations in the UGT1A1 gene.
Epidemiology The prevalence of Crigler-Najjar syndrome type 1 (CNS1) is unknown. Crigler-Najjar syndrome (CNS) has an estimated annual incidence at birth of 1/1,000,000.
Crigler-Najjar syndrome (CNS) is a hereditary disorder of bilirubin metabolism characterized by unconjugated hyperbilirubinemia due to a hepatic deficit of bilirubin glucuronosyltransferase (GT) activity. ... Differential diagnosis Differential diagnosis varies depending of the type of CNS and includes disorders of excessive bilirubin production (hemolysis) and impaired hepatic handling of bilirubin (Gilbert syndrome; see this term). Antenatal diagnosis Antenatal diagnosis is available for CNS1 and genetic counseling is recommended when parents have a family history of CNS.
MED13L haploinsufficiency syndrome is a genetic syndrome that causes intellectual disability, speech problems , and behavioral problems. People with the syndrome usually have distinctive facial features, such as eyes that slant upwards, a flat nasal bridge with a bulb-like tip, very small chin (micrognathia), large and lowset ears, and broad forehead. Most children with the syndrome have poor muscle tone (hypotonia) and may take longer to learn to sit and walk independently (delayed motor skills). Some babies with MED13L haploinsufficiency syndrome are born with heart defects, which may be mild or severe. ... There is no cure or specific treatment for MED13L haploinsufficiency syndrome. Treatment depends on the types and severity of the medical, developmental, and behavioral problems affecting the person with the syndrome and may include heart surgery and therapies such as speech, occupational , and behavioral therapy.
Family of genetic conditions caused by mutations affecting Ras genes The RASopathies are developmental syndromes caused by germline mutations (or in rare cases by somatic mosaicism ) in genes that alter the Ras subfamily and mitogen-activated protein kinases that control signal transduction , including: Capillary malformation-AV malformation syndrome Autoimmune lymphoproliferative syndrome Cardiofaciocutaneous syndrome Hereditary gingival fibromatosis type 1 Neurofibromatosis type 1 Noonan syndrome Costello syndrome , Noonan-like [1] Legius syndrome , Noonan-like Noonan syndrome with multiple lentigines , formerly called LEOPARD syndrome , Noonan-like References [ edit ] ^ Schuhmacher AJ, Guerra C, Sauzeau V, Cañamero M, Bustelo XR, Barbacid M (June 2008). "A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition" . ... External links [ edit ] RASopathies Network USA, founded 2010 rasopathiesnet.org, accessed February 27, 2014 About RAS Pathway Syndromes RASopathy Network, RAS-Pathway-syndromes.com, accessed February 27, 2014.