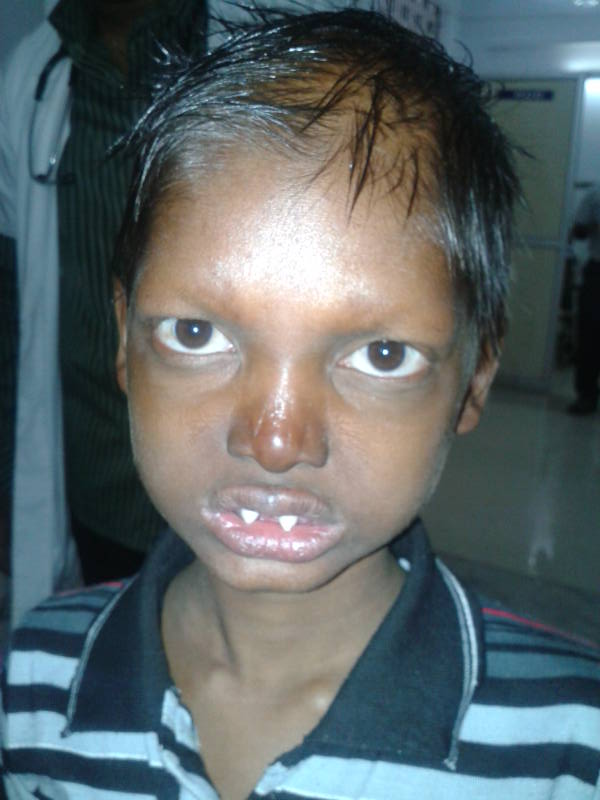
A form of mucopolysaccharidosis had earlier been designated mucopolysaccharidosis VIII (MPS VIII; MPS8), DiFerrante syndrome, or glucosamine-6-sulfate sulfatase deficiency, but the phenotype was later found to be based on incorrect data (see HISTORY). ... History Ginsburg et al. (1977) described a patient with a form of mucopolysaccharidosis that combined clinical and biochemical features of the Morquio and Sanfilippo syndromes. The patient was a 5-year-old male with short stature, mental retardation, excessive coarse hair, hepatomegaly, only mild dysostosis multiplex, and hypoplasia of the odontoid. ... Circulating lymphocytes stained with toluidine blue showed a peculiar ring-shaped metachromasia underlying the cell membrane. Unlike Morquio syndrome, cultured fibroblasts accumulated (35)S and showed delayed wash-out of (35)S. ... Ginsburg et al. (1977) suspected the existence of 2 hexosamine sulfatases, one (deficient in MPS IV, or Morquio syndrome) specific for sulfate attached to galactosamine, and one specific for sulfate attached to glucosamine.
A rare form of mucopolysaccharidosis characterized by abnormal storage of hyaluronan in lysosomes due to deficiency of hyaluronidase 1. Clinical manifestations include knee and/or hip pain associated with swelling, diffuse joint involvement with proliferative synovitis and occurrence of multiple periarticular soft-tissue masses, short stature, and dysmorphic craniofacial features (such as flattened nasal bridge, bifid uvula, and cleft palate).
Epidemiology Overall prevalence of ectodermal dysplasia syndromes is unknown, but appears rare with a presumed cumulative incidence of approximately 1/1,429. ... Although many ectodermal dysplasias are disorders with manifestations limited to the skin, hair, teeth, nails and sweat glands, multiple features of ectodermal dysplasia are accompanying signs of many syndromic conditions with systemic involvement. ... Diagnostic methods The present classification of ectodermal dysplasia syndrome is based on clinical features although a genetic classification, just as significant, has been proposed on the basis of gene function.
Ectodermal dysplasias (ED) are a group of more than 180 disorders that affect the outer layer of tissue of the embryo (ectoderm) that helps make up the skin, sweat glands, hair, teeth, and nails. Symptoms of ED can range from mild to severe and may include teeth abnormalities; brittle, sparse or absent hair; abnormal fingernails; inability to perspire ( hypohidrosis ); various skin problems; and other symptoms. Different types of EDs are caused by mutations in different genes, and can be inherited in a variety of ways. No cure currently exist for the different types of ED, but many treatments are available to address the individual symptoms.
Majeed syndrome is a rare condition characterized by recurrent episodes of fever and inflammation in the bones and skin. One of the major features of Majeed syndrome is an inflammatory bone condition known as chronic recurrent multifocal osteomyelitis (CRMO). ... Complications of congenital dyserythropoietic anemia can range from mild to severe. Most people with Majeed syndrome also develop inflammatory disorders of the skin, most often a condition known as Sweet syndrome. ... Frequency Majeed syndrome appears to be very rare; it has been reported in three families, all from the Middle East. Causes Majeed syndrome results from mutations in the LPIN2 gene.
A number sign (#) is used with this entry because of evidence that Majeed syndrome (MJDS) is caused by homozygous mutation in the LPIN2 gene (605519) on chromosome 18p11. ... Majeed et al. (2000) concluded that the combination of CRMO characterized by early onset, aggressive course, and long duration with a new microcytic type of CDA represented a new autosomal recessive syndrome. Majeed et al. (2001) reported a brother and sister, born into a consanguineous Arab family, who developed Majeed syndrome at 3 weeks and 2 months of age, respectively. ... Ferguson et al. (2005) noted that although Sweet syndrome was definitively diagnosed in only the 2 brothers reported by Majeed et al. (1989), the 2 cousins reported by Majeed et al. (1989, 2000) in that family had a history of rash that was consistent with Sweet syndrome. Ferguson et al. (2005) stated that the 2 affected sibs from the second family reported by Majeed et al. (2001) did not have Sweet syndrome, but 1 of them had a history of cutaneous pustulosis, and an obligate carrier from that family had severe psoriasis. Molecular Genetics In 2 consanguineous Arab families with Majeed syndrome, previously reported by Majeed et al. (1989, 2000, 2001), Ferguson et al. (2005) identified homozygosity for a missense mutation (S734L; 605519.0001) and a 2-bp deletion (605519.0002) in the LPIN2 gene, respectively.
Majeed syndrome is characterized by recurrent episodes of fever and inflammation in the bones and skin. ... CDA involves a shortage of red blood cells which can lead to fatigue (tiredness), weakness, pale skin, and shortness of breath. Most people with Majeed syndrome also develop inflammatory disorders of the skin, most often a condition known as Sweet syndrome. Majeed syndrome results from mutations in the LPIN2 gene.
Majeed syndrome Specialty Dermatology Majeed syndrome is an inherited skin disorder characterized by chronic recurrent multifocal osteomyelitis , congenital dyserythropoietic anemia and a neutrophilic dermatosis. [1] It is classified as an autoinflammatory bone disorder. ... The pathogenesis of this mutation with the clinical manifestations has not been elucidated. [2] See also [ edit ] TNF receptor associated periodic syndrome List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... ISBN 978-1-4160-2999-1 . ^ "Majeed syndrome" . Genetics Home Reference . Retrieved 17 April 2018 . External links [ edit ] Orphanet syndrome Majeed syndrome This dermatology article is a stub .
Epidemiology The syndrome is extremely rare. Fourteen cases born into consanguineous families, from the Middle East, India and Spain have been reported. ... Chronic recurrent multifocal osteomyelitis (CRMO) associated with Majeed syndrome is typically more severe than that of non-syndromic CRMO, and is more persistent, with short remissions and more frequent exacerbations. ... The inflammatory neutrophilic dermatosis Sweet syndrome has been reported in two patients with Majeed syndrome. ... Differential diagnosis Differential diagnoses include Caffey disease, SAPHO syndrome, non-syndromic CRMO, deficiency of the interleukin-1 receptor antagonist (DIRA) and immune deficiency. ... Nonsteroidal anti-inflammatory drugs (NSAIDs) are the main treatment options for non-syndromic CRMO, but are not likely to control bone inflammation in Majeed syndrome patients.
Majewski's polydactyly syndrome Other names Polydactyly with neonatal chondrodystrophy type I This condition is inherited in an autosomal recessive manner Majewski's polydactyly syndrome , also known as polydactyly with neonatal chondrodystrophy type I, short rib-polydactyly syndrome type II , and shorts rib-polydactyly syndrome , is a lethal form of neonatal dwarfism characterized by osteochondrodysplasia ( skeletal abnormalities in the development of bone and cartilage ) with a narrow thorax , polysyndactyly , disproportionately short tibiae , thorax dysplasia, hypoplastic lungs and respiratory insufficiency. ... "Polysyndactyly, short limbs, and genital malformations--a new syndrome?" . Zeitschrift für Kinderheilkunde . 111 (2): 118–138. doi : 10.1007/BF00446428 . ... External links [ edit ] Classification D ICD - 10 : Q77.2 OMIM : 263520 DiseasesDB : 32793 v t e Osteochondrodysplasia Osteodysplasia/ / osteodystrophy Diaphysis Camurati–Engelmann disease Metaphysis Metaphyseal dysplasia Jansen's metaphyseal chondrodysplasia Schmid metaphyseal chondrodysplasia Epiphysis Spondyloepiphyseal dysplasia congenita Multiple epiphyseal dysplasia Otospondylomegaepiphyseal dysplasia Osteosclerosis Raine syndrome Osteopoikilosis Osteopetrosis Other/ungrouped FLNB Boomerang dysplasia Opsismodysplasia Polyostotic fibrous dysplasia McCune–Albright syndrome Chondrodysplasia / chondrodystrophy (including dwarfism ) Osteochondroma osteochondromatosis Hereditary multiple exostoses Chondroma / enchondroma enchondromatosis Ollier disease Maffucci syndrome Growth factor receptor FGFR2 : Antley–Bixler syndrome FGFR3 : Achondroplasia Hypochondroplasia Thanatophoric dysplasia COL2A1 collagen disease Achondrogenesis type 2 Hypochondrogenesis SLC26A2 sulfation defect Achondrogenesis type 1B Autosomal recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia Chondrodysplasia punctata Rhizomelic chondrodysplasia punctata Conradi–Hünermann syndrome Other dwarfism Fibrochondrogenesis Short rib – polydactyly syndrome Majewski's polydactyly syndrome Léri–Weill dyschondrosteosis This article about a congenital malformation is a stub .
A rare ciliopathy with major skeletal involvement characterized by a hypoplastic thorax with short ribs and protuberant abdomen, micromelia with particularly short tibiae with ovoid configuration, pre- and postaxial polydactyly, brachydactyly, hypoplasia or aplasia of nails, and dysmorphic craniofacial features (such as prominent forehead, low-set and malformed ears, short and flat nose, lobulated tongue, micrognathia, and cleft lip/palate). Additional reported manifestations include urogenital, gastrointestinal, cardiovascular, and cerebral malformations, among others. The condition is fetal in the neonatal period.
SRTD encompasses Ellis-van Creveld syndrome (EVC) and the disorders previously designated as Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib-polydactyly syndrome (SRPS), and Mainzer-Saldino syndrome (MZSDS). ... Spranger et al. (1974) reported a similar patient (case 1) whose sib may have died of the same condition, which they referred to as Majewski type short rib-polydactyly (SRP) syndrome. They noted that polycystic kidneys occur with this condition as well as with Meckel syndrome (249000). ... The 2 patients had features typical of Mohr syndrome but, in addition, had laryngeal anomalies and hallucal and postaxial polysyndactyly of the feet typical of Majewski syndrome. In the latter condition, the oral/facial findings are almost identical to those of the Mohr syndrome. Franceschini et al. (1995) reported on a patient with manifestations typical of Mohr syndrome and of the short rib-polydactyly syndromes. ... In a later note, Urioste et al. (1994) raised the question of the syndrome being due to disruption of a gene in the 4p16 region where achondroplasia, hypochondroplasia, thanatophoric dysplasia, and Ellis-van Creveld syndrome map.
SRTD encompasses Ellis-van Creveld syndrome (EVC) and the disorders previously designated as Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib-polydactyly syndrome (SRPS), and Mainzer-Saldino syndrome (MZSDS). ... There is phenotypic overlap with the cranioectodermal dysplasias (Sensenbrenner syndrome; see CED1, 218330). For a discussion of genetic heterogeneity of short-rib thoracic dysplasia, see SRTD1 (208500). ... Saldino and Noonan (1972) suggested that this was a 'new' malformation syndrome (SRPS I) similar to EVC and ATD. ... Bernstein et al. (1985) presented 4 cases of short rib-polydactyly syndrome from 3 nonconsanguineous families. ... The authors were impressed with the similarities between type 1 ATD and short rib-polydactyly syndrome type III. Ho et al. (2000) reported a family with 2 brothers affected with mild Jeune syndrome, and a stillborn male infant, the product of a marriage between the paternal first cousin and a maternal aunt of the 2 boys, with lethal SRPS type III.
Ochoa syndrome is a disorder characterized by urinary problems and unusual facial expressions. The urinary problems associated with Ochoa syndrome typically become apparent in early childhood or adolescence. ... Frequency Ochoa syndrome is a rare disorder. About 150 cases have been reported in the medical literature. Causes Ochoa syndrome can be caused by mutations in the HPSE2 gene. ... Mutations in the HPSE2 gene that cause Ochoa syndrome result in changes in the heparanase 2 protein that likely prevent it from functioning.
A number sign (#) is used with this entry because of evidence that urofacial syndrome-2 (UFS2) is caused by homozygous or compound heterozygous mutation in the LRIG2 gene (608869) on chromosome 1p13. Description Urofacial syndrome (UFS; Ochoa syndrome) is an autosomal recessive disorder characterized by congenital urinary bladder dysfunction associated with an abnormal facial expression upon smiling, laughing, and crying. ... She also displayed the grimace-upon-smiling facies characteristic of urofacial syndrome (UFS). Despite treatment with intermittent catheterization, antimuscarinic drugs, and surgical bladder augmentation, renal failure progressed and peritoneal dialysis was planned. ... Mapping In a consanguineous Turkish family in which a girl had urofacial syndrome (UFS) and her brother had the facial features without urinary tract symptoms, Stuart et al. (2013) performed autozygosity mapping and identified a 52-Mb region of homozygosity on chromosome 1p13.2. ... Molecular Genetics In affected sibs from a consanguineous Turkish families with urofacial syndrome mapping to chromosome 1p13.2, Stuart et al. (2013) performed exome sequencing and identified homozygosity for a 1-bp deletion in the LRIG2 gene (608869.0001); the mutation segregated with disease in the family and was not found in variome databases, 116 local exomes, or 94 Turkish controls.
Summary Clinical characteristics. Urofacial syndrome (UFS) is characterized by prenatal or infantile onset of urinary bladder voiding dysfunction, abnormal facial movement with expression (resulting from abnormal co-contraction of the corners of the mouth and eyes), and often bowel dysfunction (constipation and/or encopresis). ... Diagnosis No formal diagnostic criteria for urofacial syndrome (UFS) have been published. Suggestive Findings Urofacial syndrome (UFS) should be suspected in individuals with the following clinical findings. ... Molecular Genetic Testing Used in Urofacial Syndrome View in own window Gene 1, 2 Proportion of UFS Attributed to Pathogenic Variants in Gene 3 Proportion of Pathogenic Variants 4 Detectable by Method Sequence analysis 5 Gene-targeted deletion/duplication analysis 6 HPSE2 17/25 16/17 7 1 family 8 LRIG2 4/25 4/4 9 None reported Unknown 4/25 NA 1. ... Clinical Characteristics Clinical Description The main features of urofacial syndrome (UFS) are congenital urinary bladder voiding dysfunction and an abnormality of facial movement with expression that can be observed from birth. ... Differential Diagnosis The urinary tract features of urofacial syndrome (UFS) overlap with those seen in association with multiple other conditions [Woolf et al 2014a].
A number sign (#) is used with this entry because urofacial syndrome-1 (UFS1) is caused by homozygous or compound heterozygous mutation in the HPSE2 gene (613469) on chromosome 10q24. Description The urofacial syndrome (UFS) is a rare autosomal recessive disease characterized by a severe and early-onset form of dysfunctional urinary voiding. ... Genetic Heterogeneity of Urofacial Syndrome Urofacial syndrome-2 (UFS2; 615112) is caused by mutation in the LRIG2 gene (608869) on chromosome 1p13. ... Elejalde (1979) called the disorder the Ochoa syndrome and thanked Dr. Bernardo Ochoa for referring the patients for study. ... Daly et al. (2010) reported 3 British Pakistani sibs with urofacial syndrome, born of unaffected first-cousin parents.
Ochoa syndrome is a very rare condition that causes unusual facial expressions and problems with urination. ... Other signs and symptoms may include constipation, loss of bowel control and/or muscle spasms of the anus. Ochoa syndrome can be caused by a non-working HPSE2 or LRIG2 gene and is inherited in an autosomal recessive manner.
Urofacial syndrome Other names Ochoa syndrome Ochoa Syndrome has an autosomal recessive pattern of inheritance. ... The inverted facial expression presented by children with this syndrome allows for early detection of the syndrome, which is vital for establishing a better prognosis as urinary related problems associated with this disease can cause harm if left untreated. ... This protein is believed to be altered in the case of this syndrome. [2] Studies performed on mice indicate that HPSE2 has no enzymatic activity . [5] Mutations in the HPSE2 gene on chromosome 10q23q24 have been observed to cause Ochoa Syndrome. ... "Genetic homogeneity of the urofacial (Ochoa) syndrome confirmed in a new French family". ... PMC 2933305 . PMID 20478306 . ^ "Ochoa Syndrome" . Genetics Home Reference . Retrieved 30 November 2012 .
Ochoa syndrome is characterized by the association of severe voiding dysfunction and a characteristic facial expression. ... Clinical description Patients with Ochoa syndrome present with incontinence, urinary tract infection and hydronephrosis. ... Ultrasonography, renal scan, voiding cystourethrogram and urodynamics can be used to evaluate the lower urinary tract dysfunction. Genetic counseling The syndrome is inherited as an autosomal recessive trait.
Kapur–Toriello syndrome Other names Cleft lip/palate-facial, eye, heart and intestinal anomalies syndrome Keutel syndrome has an autosomal recessive pattern of inheritance. Kapur–Toriello syndrome is a rare autosomal recessive genetic disorder. The defining feature of Kapur–Toriello syndrome is abnormal morphology of the columella, the end of the nasal septum , which in the syndrome extends below the margin of the nostrils. [1] Contents 1 Presentation 2 Causes 3 Diagnosis 4 Treatment 5 History 6 References 7 External links Presentation [ edit ] Clinical manifestations similar to all five described cases of Kapur–Toriello syndrome severe neurodevelopment delay, microphthalmia and/or coloboma , low set and malformed ears, bilateral cleft lip and palate , and constipation. [2] Out of the five cases, two cases presented with imperforated anus /rectal stenosis. ... Further Delineation of the Kapur–Toriello Syndrome. American Journal of Medical Genetics. 152A: 1013-1015. ^ Kapur, S., Toriello, H.V. (1991). Apparently new MCA/MR syndrome in sibs with cleft lip and palate and other facial, eye, heart, and intestinal anomalies.
The phenotype was compatible with Kapur-Toriello syndrome. Robin et al. (2010) reported a female infant born with multiple anomalies, including left-sided cleft lip and palate, bilateral optic nerve colobomas, ventriculoseptal defect, imperforate anus, and Dandy-Walker anomaly with cerebral ventriculomegaly. ... The distinctive columella prompted the diagnosis of Kapur-Toriello syndrome, making this the second female patient with this diagnosis to be reported. Robin et al. (2010) noted that both female patients manifested anorectal malformation, and suggested that this should be considered a component feature of the syndrome. Lefroy et al. (2015) reported a 15-year-old boy, born to nonconsanguineous parents, with complex heart defects, bilateral cleft lip and palate, unilateral clinical anophthalmia, narrow nasal bridge with bulbous tip, wide columella extending below the nares, short wide neck, profound scoliosis, cervical and thoracic hemivertebrae, and mild developmental delay. ... He also had short stature and growth hormone deficiency, features not previously reported in Kapur-Toriello syndrome. INHERITANCE - Autosomal recessive GROWTH Other - Intrauterine growth retardation HEAD & NECK Ears - Hearing loss, conductive - Low-set ears - Preauricular tags - Atresia of external auditory canal Eyes - Coloboma (iris and retina) - Cataract - Microphthalmia Nose - Bulbous nose - Long columella Mouth - Cleft lip - Cleft palate Neck - Short neck - Low posterior hairline CARDIOVASCULAR Heart - Atrial septal defect - Ventricular septal defect - Delayed closing of the ductus arteriosus ABDOMEN Gastrointestinal - Intestinal malrotation - Constipation - Anorectal malformation GENITOURINARY External Genitalia (Male) - Small penis External Genitalia (Female) - Hypoplastic labia majora Internal Genitalia (Male) - Cryptorchidism Kidneys - Heterotopic kidney SKELETAL Spine - Scoliosis Hands - Bilateral single transverse palmar creases - Finger contractures - Hypoplastic thumb - Overlapping fingers Feet - Widely spaced first and second toe - Fifth toe clinodactyly SKIN, NAILS, & HAIR Hair - Low posterior hairline NEUROLOGIC Central Nervous System - Mental retardation, severe - Seizures - Dysgenesis of the corpus callosum (1 patient) - Polymicrogyria (1 patient) - Pachygyria (1 patient) ▲ Close
Kapur-Toriello syndrome is an extremely rare syndrome characterized by facial dysmorphism, severe intellectual deficiency, cardiac and intestinal anomalies, and growth retardation.
Restless legs syndrome is a neurological condition that causes an irresistible urge to move the legs. ... Restless legs syndrome and PLMS can affect the quality and amount of sleep. ... The prevalence of restless legs syndrome increases with age. Causes Restless legs syndrome likely results from a combination of genetic and environmental factors, many of which are unknown. ... In some affected families, restless legs syndrome appears to have an autosomal dominant pattern of inheritance. ... However, the genetic changes associated with restless legs syndrome in these families have not been identified.
Genetic Heterogeneity of Restless Legs Syndrome RLS1 has been mapped to chromosome 12q. ... The authors referred to the 'painful-legs--moving-toes syndrome' in a patient whose relatives had the restless legs syndrome and proposed that the disorders are the same. ... They reported 12 pairs of monozygotic twins in which both members of 10 pairs had definite restless legs syndrome based on criteria proposed by the International Restless Legs Syndrome Study Group (IRLSSG) including the desire to move the extremities associated with paresthesia/dysesthesia; motor restlessness; worsening of symptoms at rest with temporary relief by activity; and worsening of symptoms in the evening or night. ... Trenkwalder et al. (1996) found evidence of anticipation in restless legs syndrome in 1 large German pedigree. The disorder had a 30-year age-at-onset difference between generations. ... Mapping To map genes that may play a role in the vulnerability to restless legs syndrome, Desautels et al. (2001) conducted a genomewide scan in a large French Canadian family.
Pendred syndrome Other names Goiter-deafness syndrome [1] The normal cochlea has 2 & a half turns, but, in Pendred Syndrome, there is abnormal partitioning (the central bony core is reduced in size and complexity)and a reduced number of turns leading to a Mondini cochlea which has a basal turn and a dilated apical turn (1 & a half turns). ... In some cases, language development worsens after head injury , demonstrating that the inner ear is sensitive to trauma in Pendred syndrome; this is as a consequence of the widened vestibular aqueducts usual in this syndrome. [4] Vestibular function varies in Pendred syndrome and vertigo can be a feature of minor head trauma. A goitre is present in 75% of all cases. [4] Genetics [ edit ] Pendred syndrome has an autosomal recessive pattern of inheritance. ... PMID 17622729 . ^ a b c d e Reardon W, Coffey R, Phelps PD, et al. (July 1997). "Pendred syndrome--100 years of underascertainment?" ... "Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion" .
Pendred syndrome is a disorder typically associated with hearing loss and a thyroid condition called a goiter. ... If a goiter develops in a person with Pendred syndrome, it usually forms between late childhood and early adulthood. ... Frequency The prevalence of Pendred syndrome is unknown. However, researchers estimate that it accounts for 7 to 8 percent of all hearing loss that is present from birth (congenital hearing loss). Causes Mutations in the SLC26A4 gene cause about half of all cases of Pendred syndrome. The SLC26A4 gene provides instructions for making a protein called pendrin. ... An imbalance of particular ions disrupts the development and function of the thyroid gland and structures in the inner ear, which leads to the characteristic features of Pendred syndrome. In people with Pendred syndrome who do not have mutations in the SLC26A4 gene, the cause of the condition is unknown.
A syndromic genetic deafness clinically variable characterized by bilateral sensorineural hearing loss and euthyroid goiter. Epidemiology Pendred syndrome (PDS) is one of the most frequent forms of syndromic genetic deafness. ... Genetic deafness at the DFNB4 locus is part of the phenotypic spectrum that includes PDS at one extreme and autosomal recessive non-syndromic sensorineural deafness type DFNB4 at the other. ... Differential diagnosis The differential diagnosis includes congenital cytomegaloviral infection (cCMV), BOR syndrome, and deafness at the DFNX2 locus ( POU3F4 ).
Batsakis and Nishiyama (1962) estimated that Pendred syndrome accounts for 1 to 10% of hereditary deafness. ... Peroxidase activity is normal in Pendred syndrome (Burrow et al., 1973; Ljunggren et al., 1973; Cave and Dunn, 1975). ... Reardon et al. (2000) stated that enlargement of the vestibular aqueduct, a radiologic marker, should be considered as the most likely presentation of Pendred syndrome. They found that 49 of 57 cases of deafness with enlarged vestibular aqueducts had signs of Pendred syndrome. ... Everett et al. (1997) speculated that Pendred syndrome may be more common than previously thought. ... The identification of the disease gene for Pendred syndrome prompted the need to reevaluate the syndrome to identify possible clues for the diagnosis.
Pendred syndrome is a condition usually characterized by sensorineural hearing loss in both ears (bilateral) and euthyroid goiter (enlargement of the thyroid gland with normal thyroid gland function). ... Some people have problems with balance caused by dysfunction of the part of the inner ear that helps with balance and orientation (the vestibular system). Pendred syndrome is inherited in an autosomal recessive manner.
This means that if one parent has genes that cause Lynch syndrome, there's a 50% chance that each child will have the genes that cause Lynch syndrome. ... This means the genetic changes weren't inherited. People with Lynch syndrome have the genes that cause Lynch syndrome in all the cells in their bodies. ... A negative result means gene changes that cause Lynch syndrome weren't found in your cells. It means you probably don't have Lynch syndrome. ... Treatment There's no cure for Lynch syndrome. People with Lynch syndrome often have tests to look for early signs of cancer. ... Questions could include: What causes Lynch syndrome? How is Lynch syndrome passed through families?
Genetic counseling. Lynch syndrome is inherited in an autosomal dominant manner. ... Population screening strategies for Lynch syndrome. Lynch syndrome screening guidelines for individuals with CRC have been developed by the NCCN; click here (no-fee registration and log-in required). ... Overall, a survival advantage similar to that in Lynch syndrome-related CRC has been reported in Lynch syndrome-related endometrial cancers [Maxwell et al 2001]. ... Nilbert et al [2009] determined that six of eight sarcomas in individuals with Lynch syndrome exhibited defective MMR, suggesting that sarcomas may also be part of the spectrum of Lynch syndrome tumors. ... Hamartomatous polyp syndromes. Several autosomal dominant conditions associated with an increased risk for hamartomatous polyps and CRC can usually be distinguished by their extracolonic manifestations as well as hamartomatous rather than adenomatous pathology: Juvenile polyposis syndrome (JPS), caused by pathogenic variants in SMADA4 and BMPR1A Peutz-Jeghers syndrome (PJS), caused by pathogenic variants in STK11 PTEN hamartomatous syndromes (including Cowdensyndrome and Bannayan-Riley-Ruvalcaba syndrome), caused by pathogenic variants in PTEN Hereditary diffuse gastric cancer .
Syndromic microphthalmia-12 is a genetic syndrome with the main features of small eyeballs (microphthalmia), lungs that are too small (pulmonary hypoplasia), and a defect or hole in the diaphragm that allows the abdominal contents to move into the chest cavity (diaphragmatic hernia). People with this syndrome also have progressive movement disorders that cause s evere global developmental delay. ... Facial features seen in people with this syndrome include a broad nose, and a very small chin (micrognathia). Syndromic microphthalmia-12 is caused by mutations in the RARB gene. Treatment for this syndrome is based on addressing any symptoms that a person experiences.
Diogenes syndrome Other names Senile squalor syndrome Room crammed with garbage Specialty Psychology , psychiatry Diogenes syndrome , also known as senile squalor syndrome , is a disorder characterized by extreme self-neglect , domestic squalor, social withdrawal, apathy , compulsive hoarding of garbage or animals , plus lack of shame. ... It has been shown that the syndrome is caused as a reaction to stress that was experienced by the patient. ... Diogenes Syndrome tends to occur among the elderly. ... Shah, AK (1990). "Senile squalour syndrome: what to expect and how to treat it". ... MacAnespie, H (1975). "Diogenes Syndrome". The Lancet . 305 (7909): 750. doi : 10.1016/S0140-6736(75)91664-5 .
This suggests that the condition also involves IFN dysregulation. [5] History [ edit ] The category that CANDLE syndrome is a part of, along with related disorders, falls under the banner of proteasome-associated autoinflammatory syndromes (PRAAS). ... He termed the collective symptoms Nakajo-Nishimura syndrome (NKJO). Further symptoms were added onto the overall condition from work by Nishimura, with the overall symptoms being similar to CANDLE syndrome. [3] [6] A related syndrome was described by Garg et al. in 2010 and titled Joint contractures, Muscular Atrophy, Microcytic anemia, and Panniculitis-induced Lipodystrophy (JMP) syndrome. [3] [7] The primary differences between the syndromes is the lack of fever in JMP syndrome and the lack of seizures in NKJO syndrome, both of which are present in CANDLE syndrome. [3] Though it has been proposed by Wang et al. that the different syndromes are actually just clinical phenotypic variations of the same syndrome based around different mutations of the PSMB8 gene. [8] References [ edit ] ^ a b c Tüfekçi Ö, Bengoa Ş, Karapinar TH, Ataseven EB, İrken G, Ören H (May 2015). "CANDLE syndrome: a recently described autoinflammatory syndrome". ... "Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome". Journal of the American Academy of Dermatology . ... PMID 20534754 . ^ Wang H, Das L, Tan Hung Tiong J, Vasanwala RF, Arkachaisri T (November 2014). "CANDLE syndrome: an extended clinical spectrum" .
A number sign (#) is used with this entry because of evidence that proteasome-associated autoinflammatory syndrome-1 (PRAAS1) is caused by homozygous mutation in the PSMB8 gene (177046) on chromosome 6p21. ... This disorder encompasses Nakajo-Nishimura syndrome (NKJO); joint contractures, muscular atrophy, microcytic anemia, and panniculitis-induced lipodystrophy (JMP syndrome); and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome (CANDLE). Among Japanese patients, this disorder is best described as Nakajo-Nishimura syndrome, since both Nakajo (1939) and Nishimura et al. (1950) contributed to the original phenotypic descriptions. ... Kitamura et al. (2011) provided clinical details of 3 Japanese patients with Nakajo syndrome, including the patients reported by Tanaka et al. (1993). ... Arima et al. (2011) asserted that the most striking differences between NJKO and JMP (Garg et al., 2010) were the absence of fever in JMP syndrome and the absence of seizures in NJKO.
Nakajo-Nishimura syndrome is an inherited condition that affects many parts of the body and has been described only in the Japanese population. ... The signs and symptoms of Nakajo-Nishimura syndrome overlap with those of two other conditions: one called joint contractures, muscular atrophy, microcytic anemia, and panniculitis-induced lipodystrophy (JMP) syndrome; and the other called chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. ... About 30 cases have been reported in the medical literature. Causes Nakajo-Nishimura syndrome is caused by mutations in the PSMB8 gene. ... For unknown reasons, the malfunctioning immune system triggers abnormal inflammation that can damage the body's own tissues and organs; as a result, Nakajo-Nishimura syndrome is classified as an autoinflammatory disorder. Abnormal inflammation likely underlies many of the signs and symptoms of Nakajo-Nishimura syndrome, including the nodular erythema, recurrent fevers, joint problems, and hepatosplenomegaly.
Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome (CANDLE syndrome) occurs when the immune system attacks the body by mistake. ... Since so few people have been reported with CANDLE syndrome, it is difficult to know how it affects people in the long term. CANDLE syndrome occurs when the PSMB8 gene is not working correctly. Other genes associated with CANDLE syndrome include PSMB4 , PSMA3 , and POMP .
Nakajo syndrome Other names Nodular erythema with digital changes Nakajo syndrome has an autosomal recessive pattern of inheritance . Specialty Medical genetics Nakajo syndrome , also called nodular erythema with digital changes , [1] is a rare autosomal recessive congenital disorder first reported in 1939 by A. Nakajo in the offspring of consanguineous (blood relative) parents. [2] [3] The syndrome can be characterized by erythema (reddened skin), loss of body fat in the upper part of the body, and disproportionately large eyes, ears, nose, lips, and fingers. [1] Contents 1 Genetics 2 Diagnosis 3 Treatment 4 References 5 External links Genetics [ edit ] Nakajo syndrome is inherited in an autosomal recessive manner. [3] This means the defective gene responsible for the disorder is located on an autosome , and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. ... You can help by adding to it . ( August 2017 ) References [ edit ] ^ a b Online Mendelian Inheritance in Man (OMIM): Nakajo syndrome - 256040 ^ Nakajo A (1939). "Secondary hypertrophic osteoperiostosis with pernio". ... (in Japanese). 45 : 77–86. ^ a b Kitano Y, M. E. (August 1985). "A syndrome with nodular erythema, elongated and thickened fingers, and emaciation".
There might be a discussion about this on the talk page . ( December 2011 ) ( Learn how and when to remove this template message ) TAN syndrome Specialty Dermatology Tegumental angiomyxoma-neurothekeoma ( TAN syndrome ) [1] is a syndrome , an acronym , and eponym [ citation needed ] proposed by Malaysian ophthalmologist of Chinese Descent, Tan Aik Kah (b. June 1975). [1] Angiomyxomas are associated with LAMB (lentigines, atrial myxomas, muco-cutaneous myxomas, and blue naevi) syndrome, [2] NAME (nevi, atrial myxoma, myxoid neurofibromas, and ephelides) [3] syndrome and Carney syndrome (atrial, cutaneous and mammary myxomas, lentigines, blue naevi, endocrine disorders and testicular tumours ). [4] TAN syndrome is characterized by multiple superficial angiomyxoma and neurothekeoma confined only to the skin (tegument). [1] TAN syndrome may be used to describe myxomas confined to the skin without visceral involvement. [ citation needed ] Case [ edit ] Tan et al. reported a 10-year-old girl with multiple superficial angiomyxoma associated with neurothekeoma palpebrae. [1] There was no evidence of visceral involvement. ... "Neurothekeoma palpebrae in association with multiple superficial angiomyxomas: Tegumental Angiomyxoma- Neurothekeoma syndrome (TAN syndrome)" . Clinics and Practice . 1 (3): e67. doi : 10.4081/cp.2011.e67 . ... "Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: The 'LAMB' syndrome". Journal of the American Academy of Dermatology . 10 (1): 72–82. doi : 10.1016/S0190-9622(84)80047-X .
Trauma to the median nerve or around the proximal median nerve have also been reported as causes of AIN syndrome. Although there is still controversy among upper extremity surgeons, AIN syndrome is now regarded as a neuritis (inflammation of the nerve) in most cases; this is similar to Parsonage–Turner syndrome . ... "The anterior interosseous nerve syndrome (the Kiloh-Nevin syndrome). Report and follow-up study of three cases." ... PMID 2317141 . ^ Roggenland, D, C M Heyer, M Vorgerd, and V Nicolas. "[Nervus interosseus anterior syndrome (Kiloh-Nevin syndrome)--diagnosis with MRI]." ... Ericson ^ Nigst, H, and W Dick. "Syndromes of compression of the median nerve in the proximal forearm (pronator teres syndrome; anterior interosseous nerve syndrome)." ... "Neuralgic amyotrophy; the shoulder-girdle syndrome." Lancet 1, no. 26 (June 26, 1948): 973-8.
As a result, multiple pterygium syndrome can lead to further problems with movement such as arms and legs that cannot fully extend. The two forms of multiple pterygium syndrome are differentiated by the severity of their symptoms. Multiple pterygium syndrome, Escobar type (sometimes referred to as Escobar syndrome) is the milder of the two types. Lethal multiple pterygium syndrome is fatal before birth or very soon after birth. ... Frequency The prevalence of multiple pterygium syndrome is unknown. Causes Mutations in the CHRNG gene cause most cases of multiple pterygium syndrome, Escobar type and a smaller percentage of cases of lethal multiple pterygium syndrome.
Three sibs had a lethal multiple pterygium syndrome (see 253290). Two were monozygotic twins. ... Cervical vertebral fusion, loosely labeled Klippel-Feil syndrome (118100), occurs in this disorder. This syndrome, especially in milder cases, may be confused with Noonan syndrome (163950). It is distinct from the popliteal pterygium syndrome (119500, 263650). In a study of the prune belly syndrome (100100), Welling et al. (1975) described what they considered a distinct syndrome in 2 brothers. ... Rajab et al. (2005) described 6 Omani children from 2 consanguineous families with a multiple congenital anomaly syndrome showing a high degree of phenotypic overlap with Escobar syndrome but displaying overlap with Freeman-Sheldon syndrome (193700) as well.
Both lethal (lethal and X-linked lethal multiple pterygium syndrome) and non-lethal (autosomal recessive and autosomal dominant multiple pterygium syndrome) forms occur.
A rare genetic multiple congenital anomalies/dysmorphic syndrome characterized by congenital pterygia (webbing) mainly affecting the neck and large joints, arthrogryposis multiplex, short stature, and craniofacial dysmorphism (including ptosis, downslanting palpebral fissures, high-arched palate, and retrognathia). ... The disease is a non-lethal variant of multiple pterygium syndrome.
Autosomal dominant multiple pterygium syndrome Other names Distal arthrogryposis type 8 [1] Specialty Dermatology Multiple pterygium syndrome is a cutaneous condition inherited in an autosomal dominant fashion. [2] Contents 1 Society 2 See also 3 References 4 External links Society [ edit ] Musician Patrick Henry Hughes has a type of this condition. [3] See also [ edit ] Popliteal pterygium syndrome List of cutaneous conditions Datagenno - Escobar Syndrome References [ edit ] ^ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Autosomal dominant multiple pterygium syndrome" . www.orpha.net . Retrieved 28 September 2019 . ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).
Multiple pterygium syndrome, Escobar type is characterized by webbing of skin (pterygium) and a lack of muscle movement (akinesia) that occur before birth. ... Individuals with multiple pterygium syndrome, Escobar type may also develop a restriction of the joints, known as arthrogryposis, a sideways curve of the spine (scoliosis), and distinctive facial features.
Blue diaper syndrome Other names Other Names: Hypercalcemia, familial, with nephrocalcinosis and indicanuria Blue diaper syndrome has an autosomal recessive pattern of inheritance. Medication none Blue diaper syndrome is a rare, autosomal recessive metabolic disorder characterized in infants by bluish urine-stained diapers. It is also known as Drummond's syndrome, and hypercalcemia. [1] It is caused by a defect in tryptophan absorption. ... Genetics [ edit ] Blue diaper syndrome affects males and females equally. ... PMID 11827462 . ^ RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Blue diaper syndrome" . www.orpha.net . External links [ edit ] Classification D OMIM : 211000 MeSH : C536239 DiseasesDB : 33872 External resources Orphanet : 94086 Blue diaper syndrome at NIH 's Office of Rare Diseases
Blue diaper syndrome is a rare metabolic disorder characterized by problems in the absorption of the aminoacid tryptophan and blue urine stains on diapers. ... Inheritance is autosomal recessive or X-linked recessive. Children with blue diaper syndrome may be put on a diet that restricts their intake of calcium, protein, vitamin D, and tryptophan.
Libit et al. (1972) suggested that the blue diaper syndrome can result from blue-green discoloration of the stools by a pigment elaborated by Pseudomonas aeruginosa.
Sly syndrome Other names β-glucuronidase deficiency, β-glucuronidase deficiency mucopolysaccharidosis, Sly syndrome has an autosomal recessive pattern of inheritance Specialty Endocrinology Sly syndrome , also called mucopolysaccharidosis type VII ( MPS-VII ), is an autosomal recessive lysosomal storage disease caused by a deficiency of the enzyme β-glucuronidase . ... Some people with Sly syndrome may begin to have symptoms in early childhood. ... Sly syndrome causes various musculoskeletal abnormalities that worsen with age. ... Skin cells and red blood cells of affected people will have low levels of β-glucuronidase activity. Sly syndrome can also be diagnosed through prenatal testing . [2] Treatment [ edit ] Vestronidase alfa-vjbk (Mepsevii) is the only drug approved by U.S. ... External links [ edit ] Classification D ICD - 10 : E76.2 ICD - 9-CM : 277.5 OMIM : 253220 MeSH : D016538 DiseasesDB : 8389 External resources eMedicine : ped/858 v t e Lysosomal storage diseases : Inborn errors of carbohydrate metabolism ( Mucopolysaccharidoses ) Catabolism MPS I Hurler Syndrome , Hurler-Scheie Syndrome , Scheie Syndrome MPS II: Hunter Syndrome MPS III: Sanfilippo Syndrome MPS IV: Morquio Syndrome MPS VI: Maroteaux-Lamy Syndrome MPS VII: Sly Syndrome MPS IX: Hyaluronidase deficiency
Mucopolysaccharidosis type VII (MPS VII), also known as Sly syndrome, is a progressive condition that affects most tissues and organs. ... Individuals with this condition may also have dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray. Carpal tunnel syndrome develops in many children with MPS VII and is characterized by numbness, tingling, and weakness in the hands and fingers.
A number sign (#) is used with this entry because mucopolysaccharidosis type VII (MPS7) is caused by homozygous or compound heterozygous mutation in the gene encoding beta-glucuronidase (GUSB; 611499) on chromosome 7q11. Description Mucopolysaccharidosis type VII is an autosomal recessive lysosomal storage disease characterized by the inability to degrade glucuronic acid-containing glycosaminoglycans. The phenotype is highly variable, ranging from severe lethal hydrops fetalis to mild forms with survival into adulthood. Most patients with the intermediate phenotype show hepatomegaly, skeletal anomalies, coarse facies, and variable degrees of mental impairment (Shipley et al., 1993). MPS VII was the first autosomal mucopolysaccharidosis for which chromosomal assignment was achieved.
A rare, genetic lysosomal storage disease characterized by accumulation of glycosaminoglycans in connective tissue which results in progressive multisystem involvement with severity ranging from mild to severe. The most consistent features include musculoskeletal involvement (particularly dysostosis multiplex, joint restriction, thorax abnormalities, and short stature), limited vocabulary, intellectual disability, coarse facies with a short neck, pulmonary involvement (predominantly decreased pulmonary function), corneal clouding, and cardiac valve disease. Epidemiology The prevalence at birth is reported to range between 1/345,000 -5,000,000. However, the frequency of the disease may be underestimated as the most frequent presentation is the antenatal form, which remains underdiagnosed. Clinical description Signs are extremely variable: there are prenatal forms with non-immune hydrops fetalis, and severe neonatal forms with dysmorphism, hernias, hepatosplenomegaly, club feet, dysostosis, small stature and severe hypotonia and neurological involvement that ultimately lead to profound intellectual deficit in patients who survive.
While aniridia is rarely absent in WAGR syndrome, cases have been reported without it. ... "WAGR syndrome: a clinical review of 54 cases". ... S2CID 33798707 . ^ Clericuzio CL (2004). "WAGR syndrome". In Cassidy SB, Allanson JE (eds.). Management of Genetic Syndromes (2nd ed.). New York, NY: John Wiley & Sons. ... External links [ edit ] Classification D OMIM : 194072 MeSH : D017624 DiseasesDB : 14025 External resources eMedicine : ped/2423 Orphanet : 893 International WAGR Syndrome Association DECIPHER database entry for WAGR syndrome