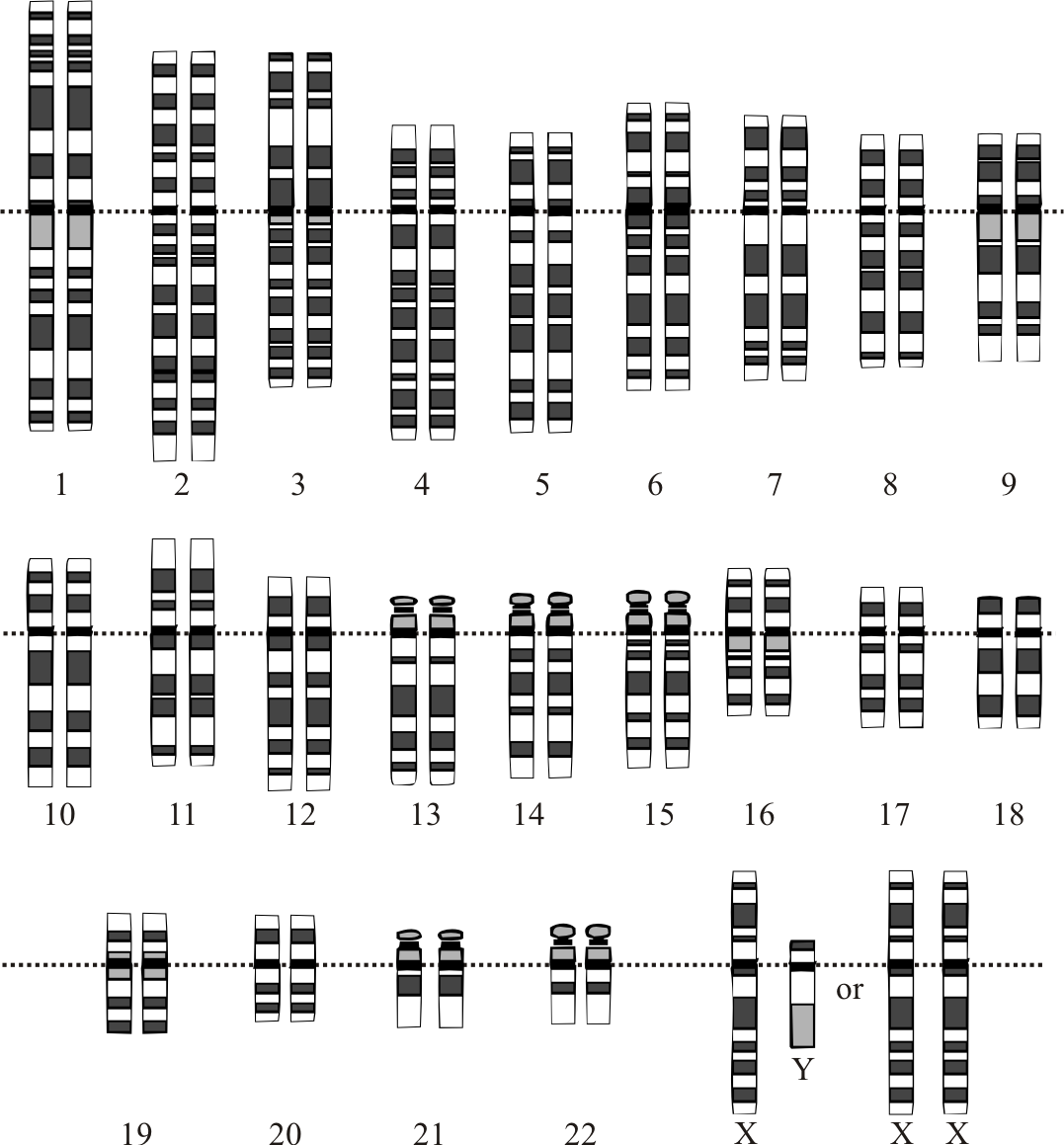
A person with the disorder may pass along either a mutated or normal copy to his or her own children. Turner syndrome Turner syndrome, a condition that affects only girls and women, results when a sex chromosome (the X chromosome) is missing or partially missing. A female inherits an X chromosome from each parent. A girl with Turner syndrome has only one fully functioning copy of the female sex chromosome rather than two. ... For example, heart problems that often occur with Turner syndrome can have a significant effect on health. An absence of sexual maturation associated with growth hormone deficiency or Turner syndrome affects both physical development and social functioning. ... Estrogen replacement therapy usually continues throughout life until women with Turner syndrome reach the average age of menopause.
Some forms of dwarfism are associated with disordered function of other organs, such as the brain or liver , sometimes severely enough to be more of an impairment than the unusual bone growth. [19] [20] Mental effects also vary according to the specific underlying syndrome. In most cases of skeletal dysplasia, such as achondroplasia, mental function is not impaired. [4] However, there are syndromes which can affect the cranial structure and growth of the brain, severely impairing mental capacity. ... It can be caused by mutations of specific genes, damage to the pituitary gland, Turner's syndrome , poor nutrition, [27] or even stress (leading to psychogenic dwarfism ). Laron syndrome (growth hormone insensitivity) is another cause. ... Other [ edit ] Other causes of dwarfism are spondyloepiphyseal dysplasia congenita , diastrophic dysplasia , pseudoachondroplasia , hypochondroplasia , Noonan syndrome , primordial dwarfism , Turner syndrome , osteogenesis imperfecta (OI), and hypothyroidism . ... "Skeletal dysplasia, growth hormone treatment and body proportion: comparison with other syndromic and non-syndromic short children" .
A number sign (#) is used with this entry because of evidence that susceptibility to epidermodysplasia verruciformis-3 (EV3) is conferred by homozygous mutation in the CIB1 gene (602293) on chromosome 15q26. Description Epidermodysplasia verruciformis-3 is characterized by onset in childhood or early adulthood of persistent disseminated flat warts and pityriasis versicolor-like lesions of the skin that are induced by cutaneous human papillomaviruses (HPVs) of the beta genus. Some patients develop nonmelanoma skin cancer, particularly on areas of the body exposed to the sun. Patients are otherwise healthy and normally resistant to other microorganisms, including other viruses and skintropic pathogens, and even all other cutaneous and mucosal HPVs (de Jong et al., 2018). For a discussion of genetic heterogeneity of susceptibility to epidermodysplasia verruciformis, see EV1 (226400).
In addition, an acquired epidermodysplasia verruciformis-like syndrome has been described in patients with impaired cell-mediated immunity, mainly HIV-infected subjects.
A number sign (#) is used with this entry because of evidence that susceptibility to epidermodysplasia verruciformis-2 (EV2) is conferred by homozygous mutation in the TMC8 gene (605829) gene on chromosome 17q25. Description Epidermodysplasia verruciformis (EV) is a rare genodermatosis associated with a high risk of skin cancer. EV results from an abnormal susceptibility to specific related human papillomavirus (HPV) genotypes and to the oncogenic potential of some of them, mainly HPV5. Infection with EV-associated HPV leads to the early development of disseminated flat wart-like and pityriasis versicolor-like lesions. Patients are unable to reject their lesions, and cutaneous Bowen carcinomas in situ and invasive squamous cell carcinomas develop in about half of them, mainly on sun-exposed areas (summary by Ramoz et al., 2000).
A number sign (#) is used with this entry because of evidence that susceptibility to epidermodysplasia verruciformis-1 (EV1) is conferred by homozygous or compound heterozygous mutation in the TMC6 gene (605828) on chromosome 17q25. Description Epidermodysplasia verruciformis (EV) is a rare genodermatosis associated with a high risk of skin cancer. EV results from an abnormal susceptibility to specific related human papillomavirus (HPV) genotypes and to the oncogenic potential of some of them, mainly HPV5. Infection with EV-associated HPV leads to the early development of disseminated flat wart-like and pityriasis versicolor-like lesions. Patients are unable to reject their lesions, and cutaneous Bowen carcinomas in situ and invasive squamous cell carcinomas develop in about half of them, mainly on sun-exposed areas (summary by Ramoz et al., 2000).
Androphy et al. (1985) described a kindred in which a 56-year-old man had EDV, none of his 5 sons or 5 daughters had EDV, and 4 of his grandsons (through 2 daughters) had EDV. All were infected with human papillomavirus 3 (HPV 3) and with HPV 8. The proband, who had onset of warts at age 5 years with no regression over the next 50 years and with extension to cover about 10% of his skin surface, had squamous carcinoma arising on sun-exposed areas of the face, ears, neck, back, arms, and hands over the previous 25 years. Other pedigrees have suggested autosomal inheritance although whether dominant as suggested by some families or recessive as suggested by parental consanguinity (see 226400) is not certain. Inheritance - X-linked form Skin - Epidermodysplasia verruciformis - Human papillomavirus (HPV 3 and HPV 8) infection - Warts - Squamous skin carcinoma ▲ Close
A number sign (#) is used with this entry because of evidence that susceptibility to epidermodysplasia verruciformis-5 (EV5) is conferred by homozygous mutation in the IL7 gene (146660) on chromosome 8q21. One such family has been reported. Description Epidermodysplasia verruciformis-5 is an autosomal recessive immunologic disorder characterized by onset of warts and verrucous or plaque-like skin lesions associated with HPV infection. Immunologic workup shows T-cell lymphopenia, particularly affecting CD4+ T cells. There is an increased risk of skin malignancy, and some patients may have other symptoms of immune dysfunction (summary by Horev et al., 2015). For a discussion of genetic heterogeneity of susceptibility to epidermodysplasia verruciformis, see EV1 (226400).
Contents 1 History 1.1 Maternal irradiation and congenital malformations 1.2 Congenital rubella syndrome (CRS) 1.3 Thalidomide Tragedy (1950) 2 Testing and Risk assessment 3 Toxicity effects 3.1 Effects on Neurulation 3.2 Fetal alcohol syndrome (FAS) 3.3 DES (diethylstilbestrol) 3.4 Methylmercury 3.5 Chlorpyrifos 3.6 Environmental Endocrine disruptors 3.7 Epigenetics 4 Major Developmental Toxicants 5 References 6 Sources History [ edit ] Researchers have been able to ascertain toxicity associated with abnormal development with new breakthrough in developmental biology . ... Cataracts in a child's eyes due to Congenital Rubella Syndrome (CRS). Congenital rubella syndrome (CRS) [ edit ] Main article: Congenital rubella syndrome Rubella was the first recognized human epidemic of malformations. ... This was the first published recognition of congenital rubella syndrome (CRS). The progeny had congenital eye, heart and ear defects as well as mental retardation. [5] Thalidomide Tragedy (1950) [ edit ] Main article: Thalidomide Thalidomide was extensively used for the treatment of nausea in pregnant women in the late 1950s and early 1960s until it became apparent in the 1960s that it resulted in severe birth defects. ... It is the process of formation of a flat neural plate which then convolutes to form the hollow neural tube. [8] It is considered to be one of the main targets of developmental toxicity and defects in neurulation is a common consequence of toxicant exposure and results in large proportion of human defects. [9] Facial characteristics that suggest the diagnosis of FAS. Fetal alcohol syndrome (FAS) [ edit ] Main article: Fetal alcohol spectrum disorder Fetal alcohol spectrum disorders (FASD) is a term that constitutes the set of conditions that can occur in a person whose mother drank alcohol during the course of pregnancy. ... Recognizing and Managing Children with Fetal Alcohol Syndrome/Fetal Alcohol Effects: A Guidebook . ^ "Home | FASD | NCBDDD | CDC" . www.cdc.gov .
. ^ a b c d e f g h i j k l m Hip Dislocation Treatment & Management at eMedicine External links [ edit ] Classification D ICD - 10 : S73.0 , Q65.0 - Q65.2 ICD - 9-CM : 835 OMIM : 142700 MeSH : D006618 DiseasesDB : 3056 External resources eMedicine : emerg/144 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum v t e Dislocations / subluxations , sprains and strains Joints and ligaments Head and neck Dislocation of jaw Whiplash Shoulder and upper arm GH ( Dislocated shoulder ) AC ( Separated shoulder ) ALPSA lesion SLAP tear Bankart lesion Elbow and forearm Pulled elbow Gamekeeper's thumb Hip and thigh Hip dislocation Knee and leg Tear of meniscus Anterior cruciate ligament injury Unhappy triad Patellar dislocation Knee dislocation Ankle and foot Sprained ankle ( High ankle sprain ) Turf toe Muscles and tendons Shoulder and upper arm Rotator cuff tear Hip and thigh Pulled hamstring Knee and leg Patellar tendon rupture Achilles tendon rupture Shin splints
Overview Presbyopia is the gradual loss of your eyes' ability to focus on nearby objects. It's a natural, often annoying part of aging. Presbyopia usually becomes noticeable in your early to mid-40s and continues to worsen until around age 65. You may become aware of presbyopia when you start holding books and newspapers at arm's length to be able to read them. A basic eye exam can confirm presbyopia. You can correct the condition with eyeglasses or contact lenses. You might also consider surgery. Symptoms Presbyopia develops gradually.
This condition is also a medical emergency. HELLP syndrome. HELLP syndrome — which stands for hemolysis (destruction of red blood cells), elevated liver enzymes and low platelet count — can rapidly become life-threatening. Symptoms of hemolysis elevated liver enzymes and low platelet count (HELLP) syndrome include nausea and vomiting, headache, and upper right abdominal pain. HELLP syndrome is particularly dangerous because it represents damage to several organ systems.
Among 6 offspring of first-cousin parents, Inbal et al. (1995) identified a brother and sister with a seemingly 'new' syndrome characterized by myopathy, sideroblastic anemia, lactic acidosis, mental retardation, microcephaly, high palate, high philtrum, distichiasis, and micrognathia. ... However, examination of DNA from the affected sibs showed no deletions in the mitochondrial DNA and no mutations of the type identified in the syndrome of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS; 540000) or the syndrome of myoclonus and epilepsy associated with ragged-red fibers (MERRF; 545000). ... The younger brother, who was still alive at 13 years of age, had a much milder neuromuscular syndrome with no muscle wasting, and did not require transfusions for his sideroblastic anemia; however, he had cognitive and behavioral abnormalities with moderate mental retardation, hyperactivity, and panic attacks.
Electrocardiogram (EKG) confirmed Wolff-Parkinson-White syndrome (194200). At 6 years of age, the patient was nonambulatory and nonverbal; height and weight were below the 3rd percentile.
A number sign (#) is used with this entry because of evidence that myopathy, lactic acidosis, and sideroblastic anemia-2 (MLASA2) is caused by homozygous mutation in the YARS2 gene (610957) on chromosome 12p11. Description Myopathy, lactic acidosis, and sideroblastic anemia-2 is an autosomal recessive disorder of the mitochondrial respiratory chain. The disorder shows marked phenotypic variability: some patients have a severe multisystem disorder from infancy, including cardiomyopathy and respiratory insufficiency resulting in early death, whereas others present in the second or third decade of life with sideroblastic anemia and mild muscle weakness (summary by Riley et al., 2013). For a discussion of genetic heterogeneity of MLASA, see MLASA1 (600462). Clinical Features Sasarman et al. (2002) reported a 34-year-old man of Lebanese descent (patient E) with a lifelong history of muscle weakness, exercise intolerance, occasional muscle cramping and stiffness after running, and small muscles.
Mitochondrial myopathy and sideroblastic anemia belongs to the heterogeneous family of metabolic myopathies. It is characterised by progressive exercise intolerance manifesting in childhood, onset of sideroblastic anaemia around adolescence, lactic acidaemia, and mitochondrial myopathy. Epidemiology Less than 10 cases have been described so far. Etiology A 656C-->T mutation in the nuclear pseudouridine synthase 1 gene ( PUS1 ), localised to 12q24.33, has recently been identified in some patients. Deficient pseudouridylation of mitochondrial tRNAs may be responsible for the oxidative phosphorylation disorder. Diagnostic methods Muscle biopsy demonstrates low activity of complexes 1 and 4 of the respiratory chain and paracrystalline inclusions can be revealed in most mitochondria by electron microscopy.
A number sign (#) is used with this entry because X-linked severe congenital neutropenia (SCNX) is caused by hemizygous mutation in the WAS gene (300392) on chromosome Xp11. See also Wiskott-Aldrich syndrome (WAS; 301000), an allelic disorder. ... Clinical Features In a 3-generation family of European descent, Devriendt et al. (2001) described 5 males who presented with a novel X-linked immunodeficiency syndrome characterized by recurrent major bacterial infections, severe congenital neutropenia, and monocytopenia. ... No evidence for linkage with 19p13.3 was found; evidence pointing to linkage to the proximal Xp region containing the Wiskott-Aldrich syndrome locus was found. Molecular Genetics Mutation analysis of the WAS gene by Devriendt et al. (2001) revealed a missense mutation (L270P; 300392.0012) in all affected males and carrier females. ... INHERITANCE - X-linked recessive SKIN, NAILS, & HAIR Skin - No eczema HEMATOLOGY - Severe congenital neutropenia - Low to low-normal platelet count - Normal mean platelet volume (MPV) IMMUNOLOGY - Increased activated CD8+ T cells - Decreased CD4+/CD8+ ratio - Recurrent major bacterial infections - Decreased CD3(-)CD16/15(+) natural killer cells - Low-normal IgA levels MISCELLANEOUS - Allelic to Wiskott-Aldrich syndrome ( 301000 ) and X-linked thrombocytopenia ( 313900 ) MOLECULAR BASIS - Caused by mutation in the WAS gene (WAS, 300392.0012 ) ▲ Close
X-linked severe congenital neutropenia is an immunodeficiency syndrome characterized by recurrent major bacterial infections, severe congenital neutropenia, and monocytopenia.
There is a known association with Goldenhar syndrome (oculo-auriculo-vertebral syndrome) [9] and with Wildervanck syndrome . [10] [11] [12] There may also be an association with congenital cartilaginous rest of the neck . [ citation needed ] Management [ edit ] Simple surgical excision is curative. [13] The recommended treatment is that the skin is peeled off the extra-auricular tissue and protruding cartilage remnants are trimmed. [14] Normal appearance is achieved in majority of cases. ... "Accessory tragi and associated syndromes involving the first branchial arch".
Ventricular lead dislodgement is less common compared to atrial lead dislodgement. [2] Causes Twiddler's Syndrome The patient's constant manipulation of the pulse generator within its skin pocket can lead to a dislodgement of the device. [6] The generator is rotated on its longitudinal axis, which causes traction and results in a lead dislodgement. [5] Reel's Syndrome Like Twiddler's Syndrome, it is the manipulation of the pulse generator, but instead the generator is rotated on its transverse axis, which rolls the lead around the generator, creating dislodgement. [5] Direct trauma over the system. [5] Lead fracture [2] Unit malfunction Battery failure, component malfunction, or generator failure [4] Problems at the insertion site Infection of the insertion site can cause local inflammation or the formation of an abscess in the pulse generator pocket. [2] Infection can cause the erosion of part of the pacing system that is in the skin. [2] Failures related to exposure to high voltage electricity or high intensity microwaves [4] Indirect factors [ edit ] Power-generating equipment, arc welding equipment and powerful magnets (as in medical devices, heavy equipment or motors) can inhibit pulse generators. ... PMID 16943923 . ^ Salahuddin, Mohammad; Cader, Fathima Aaysha; Nasrin, Sahela; Chowdhury, Mashhud Zia (2016-01-01). "The pacemaker-twiddler's syndrome: an infrequent cause of pacemaker failure" .
Kikuchi-Fujimoto disease (KFD) is a benign and self-limited disorder, characterized by regional cervical lymphadenopathy with tenderness, usually accompanied with mild fever and night sweats. Less frequent symptoms include weight loss, nausea, vomiting, sore throat. Epidemiology Kikuchi-Fujimoto disease is an extremely rare disease known to have a worldwide distribution with higher prevalence among Japanese and other Asiatic individuals. Prevalence is unknown. Only isolated cases are reported in Europe. Etiology The clinical, histopathological and immunohistochemical features appear to point to a viral etiology, a hypothesis that still has not been proven. Diagnostic methods KFD is generally diagnosed on the basis of an excisional biopsy of affected lymph nodes.
Kikuchi disease is a benign (non-cancerous) condition of the lymph nodes. The main symptoms include swollen lymph nodes in the neck, mild fever , and night sweats . Less common symptoms include weight loss, nausea, vomiting, and sore throat. While the exact cause of this condition is unknown, infectious and autoimmune causes have been suggested. Kikuchi disease usually gets better (resolves) on its own within one to four months (although it may take up to a year), with or without treatment.
A number sign (#) is used with this entry because of evidence that Alkuraya-Kucinskas syndrome (ALKKUCS) is caused by homozygous or compound heterozygous mutation in the KIAA1109 gene (611565) on chromosome 4q27.
It also occurs more often in people who also have: Marfan syndrome Ehlers-Danlos syndrome Osteogenesis imperfecta Noonan syndrome Turner syndrome Complications Severe cases of pectus excavatum can compress the heart and lungs or push the heart over to one side.
A small sample size test found in least some cases, 37% of individuals have an affected first degree family member. [9] As of 2012, a number of genetic markers for pectus excavatum have also been discovered. [10] It was believed for decades that pectus excavatum is caused by an overgrowth of costal cartilage, however people with pectus excavatum actually tend to have shorter, not longer, costal cartilage relative to rib length. [11] Pectus excavatum can be present in other conditions too, including Noonan syndrome , Marfan syndrome [12] and Loeys–Dietz syndrome as well as other connective tissue disorders such as Ehlers–Danlos syndrome . [13] Many children with spinal muscular atrophy develop pectus excavatum due to their diaphragmatic breathing . ... PMID 30691856 . ^ "eMedicine — Marfan Syndrome" . Harold Chen . 2018-05-23. ^ Creswick HA1, Stacey MW, Kelly RE Jr, Gustin T, Nuss D, Harvey H, Goretsky MJ, Vasser E, Welch JC, Mitchell K, Proud VK (October 2006). ... PMID 22394989 . ^ Non-surgical sunken chest treatment device may eliminate surgery , Mass Device, November 2012 ^ Raver-Lampman (November 2012), First patients in US receive non-surgical device of sunken chest syndrome , AAAS ^ a b Lopez M, Patoir A, Costes F, Varlet F, Barthelemy JC, Tiffet O (2016). ... Elsevier Masson SAS; 2012 Aug 8:1–6. ^ "Pectus Excavatum & Poland Syndrome treatment I AnatomikModeling" . ^ J-P. ... Pectus excavatum at Curlie v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum Authority control GND : 4186076-7