Affected individuals have neurologic symptoms, including sensorineural deafness. Another allelic disorder, Arts syndrome (301835), results from loss of PRPS1 activity and has a severe neurologic phenotype including mental retardation, early-onset hypotonia, and susceptibility to infections. ... All 3 had facial stigmata suggestive of a genetic syndrome, including triangular face with prominent forehead, epicanthus, hypotelorism, beaked nose, broad mouth, and hyperopia. ... Moran et al. (2012) described a patient with PRPS1 superactivity as well as features of Arts syndrome (301835), a developmental disorder also caused by mutations in the PRPS1 gene. ... In addition, he had recurrent infections and early death at age 27 months from infection, consistent with Arts syndrome. A maternal uncle with similar symptoms had died of pneumonia at age 2. ... In a patient with a complex phenotype comprising Arts syndrome and PRPS1 superactivity, Moran et al. (2012) detected a missense mutation in the transversion in exon 4 of the PRPS1 gene (V142L; 311850.0017).
Computer-assisted molecular modeling has shown that pathogenic variants causing Arts syndrome and CMTX5 disturb the ATP binding site of PRS-I. ... Disorders of purine and pyrimidine metabolism that overlap with PRPS1- related disorders are hypoxanthine-guanine phosphoribosyltransferase (HPRT; EC 2.4.2.8) deficiency (see also Lesch-Nyhan Syndrome) and S-adenosylhomocysteine hydrolase (AHCY) deficiency [Baric et al 2004]. ... Prevention of Secondary Complications By analogy to individuals with Lesch-Nyhan syndrome, who also have marked purine overproduction, xanthine oxidase inhibition with allopurinol or febuxostat may result in the formation of xanthine urinary tract stones. ... For an open-label clinical trial of SAM in two Australian brothers (from ages 14 and 13 in 2010) with Arts syndrome [Christodoulou et al, unpublished data] (approved by the ethics and drug committees, Children's Hospital at Westmead, Sydney, Australia), oral SAM supplementation was set at 30 mg/kg/day. ... Mildly affected heterozygous females from families with Arts syndrome may also benefit from SAM supplementation in their diet, although this remains to be tested.
Phosphoribosylpyrophosphate synthetase superactivity (PRS superactivity) is characterized by the overproduction and accumulation of uric acid (a waste product of normal chemical processes) in the blood and urine. The overproduction of uric acid can lead to gout, which is arthritis caused by an accumulation of uric acid crystals in the joints. Individuals with PRS superactivity also develop kidney or bladder stones that may result in episodes of acute kidney failure. There are two forms of PRS superactivity, a severe form that begins in infancy or early childhood, and a milder form that typically appears in late adolescence or early adulthood. In both forms, a kidney or bladder stone is often the first symptom. Gout and impairment of kidney function may develop if the condition is not adequately controlled with medication and dietary restrictions.
Loss of function mutations in PRPS1 are linked to Charcot-Marie-Tooth X type 5, X-linked non-syndromic sensorineural deafness and Arts syndrome, together these diseases form part of a spectrum of PRPS1 -related disorders.
A severe form of phosphoribosylpyrophosphate (PRPP) synthetase superactivity, an X-linked disorder of purine metabolism, characterized by early onset hyperuricemia and hyperuricosuria, and clinically manifesting with urolithiasis, gout and neurodevelopmental anomalies consisting of variable combinations of sensorineural hearing loss, hypotonia, and ataxia. Epidemiology PRPP synthetase superactivity is a rare disorder with 30 families described in the literature. The severe form accounts for approximately 25% of cases. The disorder predominantly affects males. Clinical description The phenotype varies greatly among patients. The severe form manifests in infancy or early childhood (but may occur earlier) usually with uric acid crystalluria and urinary stones (kidney and/or bladder), followed by the development of gouty arthritis and eventually renal failure as a result of obstructive uropathy from uric acid crystal deposition. This form also shows neurologic impairment, mainly sensorineural hearing loss, hypotonia, ataxia, developmental delay, and /or intellectual disability.
A mild form of phosphoribosylpyrophosphate (PRPP) synthetase superactivity, an X-linked disorder of purine metabolism, characterized by adolescent or early adult-onset hyperuricemia and hyperuricosuria, leading to urolithiasis and gout. Epidemiology PRPP synthetase superactivity is a rare disorder with 30 families described in the literature. The mild form accounts for approximately 75% of cases. The disorder predominantly affects males. Clinical description Mild PRPP synthetase superactivity typically manifests in late adolescence or early adulthood, but may manifest earlier, with uric acid crystalluria and urinary stones (kidney and/or bladder), followed by the development of gouty arthritis and eventually renal failure as a result of obstructive uropathy from uric acid crystal deposition. This form is not associated with any neurodevelopmental anomalies. Etiology The disease is due to overactivity of PRPP synthetase 1 (PRS-I), an enzyme that catalyzes the synthesis of PRPP, a cofactor involved in the synthesis of purine and pyrimidine nucleotides.
Of the 10 affected males in 4 generations in this family, all except this patient died in the first months of life. Congenital Short Bowel Syndrome Kern et al. (1990) reported a nonconsanguineous Italian family in which 3 male sibs were born with a short small bowel, malrotation, and functional bowel obstruction. ... Siva et al. (2002) reported a 35-year-old male with synovial lipomatosis and congenital short bowel syndrome. At 15 years of age the length of his small intestine was found to be 90 inches, about one-third of normal. ... Many of these features overlapped with that of the MECP2 duplication syndrome (300260). Clayton-Smith et al. (2009) also commented on a facial phenotype associated with Xq28 duplications: a narrow pinched appearance of the nose, deep-set eyes, prominent chin, small everted lower lip, and flat nasal bridge. ... They noted that, different from the male with PVNH and constipation described by Hehr et al. (2006) (see 300017.0024), the phenotype in the family originally described by Auricchio et al. (1996) was most distinguished by severe CIIP, present at birth, that was lethal unless promptly corrected by surgery. Congenital Short Bowel Syndrome Van der Werf et al. (2013) reported that the sibs reported by Kern et al. (1990) and the unrelated singleton reported by Siva et al. (2002) with X-linked congenital short bowel syndrome all had the same 2-basepair deletion in FLNA (300017.0035).
Clinical Features Roy et al. (1980) reported a father and 3 sons with chronic idiopathic intestinal pseudoobstruction who had multiple bouts of abdominal colic, abdominal distention, and diarrhea. Histologic examination of ileal and colonic tissue revealed abnormalities in the myenteric plexus, including hyperplastic nerve trunks and ganglion cells, as well as an increased number of ganglion cells. Silver staining showed striking changes in the neurons, some of which were swollen, distorted, and vacuolated; there were also several abnormal, hypertrophied axons. Neurologic examination, including pupillary reflexes, was normal. Mayer et al. (1986) described a family with visceral neuropathy without extraintestinal manifestations transmitted over at least 4 generations in an autosomal dominant manner. A characteristic dilation of the jejunum and ileum was seen radiographically in 7 affected family members.
Tanner et al. (1976) described 3 infants with functional intestinal obstruction, short small intestine, malrotation, and pyloric hypertrophy. In all 3, absence of ongoing peristalsis could be related to failure of development of the argyrophil myenteric plexus. They identified 4 previously reported infants with similar symptoms. Affected sibs and parental consanguinity indicated autosomal recessive inheritance. Thickening of the bowel wall, a striking and diagnostic feature at laparotomy, may be 'work hypertrophy' from the stretching of the bowel. Diagnosis is unlikely without laparotomy, which is indicated because of the genetic implications and because prolonged intravenous nutrition is not indicated.
Chronic intestinal pseudo-obstruction (CIPO) is a rare gastrointestinal motility disorder characterized by recurring episodes resembling mechanical obstruction in the absence of organic, systemic, or metabolic disorders, and without any physical obstruction being detected by X-ray or during surgery. CIPO develops predominantly in children and may be present at birth. Epidemiology The prevalence remains unknown. The male-to-female ratio is about 1.5:1 to 4:1. Clinical description Patients commonly present with severe chronic "obstructive" symptoms: abdominal pain, distension/fullness, nausea/vomiting, diarrhea and/or intractable constipation, malabsorption of nutrients leading to weight loss and/or failure to thrive. Laboratory abnormalities usually reflect the degree of malabsorption and malnutrition. The radiological findings commonly include paralytic ileus or signs of apparent clinical obstruction with dilated loops of bowel.
Chronic intestinal pseudo-obstruction (CIPO) is a rare disease characterized by repetitive episodes or continuous symptoms of bowel obstruction when no blockage exists. Problems with nerves, muscles, or interstitial cells of Cajal (the cells that set the pace of intestinal contractions) prevent normal contractions and cause problems with the movement of food, fluid, and air through the intestines. The most common symptoms are abdominal swelling or bloating (distention), vomiting, abdominal pain, failure to thrive, diarrhea, constipation, feeding intolerance and urinary symptoms. CIPO can occur in people of any age. It may be primary or secondary. Primary or idiopathic (where the cause is unknown) CIPO occurs by itself. Secondary CIPO develops as a complication of another medical condition.
As pointed out by Barone et al. (1996), Fadda et al. (1983) differentiated 2 clinical and histochemical forms of neuronal intestinal dysplasia, NID A (see 243180) and NID B. NID A is a very rare condition characterized by congenital hypoplasia or aplasia of the sympathetic innervation of the intestine. In NID B, the entity discussed in this entry, the parasympathetic submucous plexus is primarily affected. As indicated by Borchard et al. (1991), characteristic histologic and histochemical findings, on which the diagnosis can be based, are (1) hyperplasia of submucosal plexuses with giant submucosal ganglia, (2) increased acetylcholinesterase activity in nerve fibers around submucosal blood vessels, (3) increased acetylcholinesterase activity in nerve fibers of the lamina propria mucosae, and (4) heterotopic ganglion cells in the lamina propria mucosae and in the muscularis mucosae. While most cases of NID B are sporadic, the observation of the few familial clusters suggest autosomal dominant inheritance (Scharli (1992)).
Autopsy showed no evidence of limb anomalies associated with small patella syndrome. Lungs had 3 lobes bilaterally and features of congenital alveolar dysplasia thought to represent growth arrest in the late canalicular/early saccular phase of lung development. ... He had no obvious dysmorphic features, but radiographs were not available to assess for small patella syndrome. Echocardiogram showed a large secundum atrial septal defect. ... The mother and one daughter had LADD syndrome (149730). The other daughter (P040) had pulmonary hypoplasia and died at day 5 of age after intracranial hemorrhage on ECMO. She had a broad forehead but no other features of LADD syndrome. A second family had a father and daughter with LADD syndrome and another daughter (P076) who died at 5 days of age due to pulmonary hypoplasia.
Primary pulmonary hypoplasia is a rare, isolated, genetic developmental defect during embryogenesis characterized by congenital malformation of pulmonary parenchyma with absence of other anomalies. Neonatally patients present with decreased breath sounds, small lung volume and severe respiratory distress that is not responsive to aggressive treatment (including surfactant instillation/ mechanical respiratory support). It is usually not compatible with life.
Clinical Features Finer et al. (1978) reported 2 male sibs with several congenital anomalies suggestive of the VATER association with prominent features of a caudal regression syndrome (see 600145). The older infant had multiple cardiac abnormalities, including transposition of the great arteries and ventricular septal defect. ... Of these 76 cases, 7 had recognized causes (5 chromosomal anomalies and 2 single-gene disorders), and another 19 had recognized clinical phenotypes or syndromes of unknown etiology. Of the remaining 50 cases, which were considered to have VACTERL association, ventricular septal defect was the most common cardiovascular defect (30%) and renal agenesis was the most common renal anomaly (30%); the most common limb defects were reduction deformities (34%) and polydactyly (20%). ... Killoran et al. (2000) noted phenotypic overlap between the VATER/VACTERL association and what had been called PIV syndrome as described by Say and Gerald (1968), Filippi (1972), and Kaufman et al. (1972). Overlapping features included polydactyly (central or postaxial), shortened fingers, hypoplastic nails, renal anomalies, and imperforate anus. They viewed PIV syndrome as a historic antecedent to the VATER/VACTERL association. ... Castori et al. (2008) concluded that the relatively high frequency of tibial ray anomalies in VACTERL patients reflected the principle of homology of the developmental field theory. Townes-Brocks syndrome (107480) and MURCS association (601076) include some of the same features as VATER association.
VACTERL association is a non-random association of birth defects that affects multiple parts of the body. The term VACTERL is an acronym with each letter representing the first letter of one of the more common findings seen in affected individuals: (V) = vertebral abnormalities; (A) = anal atresia ; (C) = cardiac (heart) defects; (T) = tracheal anomalies including tracheoesophageal (TE) fistula ; (E) = esophageal atresia ; (R) = renal (kidney) and radial (thumb side of hand) abnormalities; and (L) = other limb abnormalities. Other features may include (less frequently) growth deficiencies and failure to thrive; facial asymmetry ( hemifacial microsomia ); external ear malformations; intestinal malrotation ; and genital anomalies. Intelligence is usually normal. The exact cause of VACTERL association is unknown; most cases occur randomly, for no apparent reason. In rare cases, VACTERL association has occurred in more than one family member.
Contents 1 Signs and symptoms 2 Diagnosis 2.1 Differential diagnosis 3 Prognosis 4 History 4.1 Disease or syndrome 5 Notable cases 6 References 7 External links Signs and symptoms [ edit ] As a result of lower motor neuron degeneration, the symptoms of PMA include: atrophy fasciculations muscle weakness Some patients have symptoms restricted only to the arms or legs (or in some cases just one of either). ... The name "spinal muscular atrophy" is ambiguous as it refers to any of various spinal muscular atrophies , including the autosomal recessive spinal muscular atrophy caused by a genetic defect in the SMN1 gene. Disease or syndrome [ edit ] Since its initial description in 1850, there has been debate in the scientific literature over whether PMA is a distinct disease with its own characteristics, or if lies somewhere on a spectrum with ALS , PLS , and PBP . ... "The history of progressive muscular atrophy: Syndrome or disease?". Neurology . 70 (9): 723–727. doi : 10.1212/01.wnl.0000302187.20239.93 . ... External links [ edit ] Classification D ICD - 9-CM : 335.21 MeSH : D009134 DiseasesDB : 29149 v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis
A rare motor neuron disease characterized by isolated lower motor neuron features, including progressive flaccid weakness, muscle atrophy, fasciculations, and reduced or absent tendon reflexes. Onset is in late adulthood, with men being affected more often than women. Upper motor neuron signs may develop later in some cases. Occurrence of respiratory insufficiency determines the prognosis. Neuropathological analysis shows intraneuronal Bunina bodies and ubiquitin-positive inclusions.
External links [ edit ] Classification D ICD - 10 : I27.2 OMIM : 265450 MeSH : C14.907.690 External resources Orphanet : 31837 Scholia has a topic profile for Pulmonary venoocclusive disease . v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline v t e Cardiovascular disease (vessels) Arteries , arterioles and capillaries Inflammation Arteritis Aortitis Buerger's disease Peripheral artery disease Arteriosclerosis Atherosclerosis Foam cell Fatty streak Atheroma Intermittent claudication Critical limb ischemia Monckeberg's arteriosclerosis Arteriolosclerosis Hyaline Hyperplastic Cholesterol LDL Oxycholesterol Trans fat Stenosis Carotid artery stenosis Renal artery stenosis Other Aortoiliac occlusive disease Degos disease Erythromelalgia Fibromuscular dysplasia Raynaud's phenomenon Aneurysm / dissection / pseudoaneurysm torso : Aortic aneurysm Abdominal aortic aneurysm Thoracic aortic aneurysm Aneurysm of sinus of Valsalva Aortic dissection Aortic rupture Coronary artery aneurysm head / neck Intracranial aneurysm Intracranial berry aneurysm Carotid artery dissection Vertebral artery dissection Familial aortic dissection Vascular malformation Arteriovenous fistula Arteriovenous malformation Telangiectasia Hereditary hemorrhagic telangiectasia Vascular nevus Cherry hemangioma Halo nevus Spider angioma Veins Inflammation Phlebitis Venous thrombosis / Thrombophlebitis primarily lower limb Deep vein thrombosis abdomen Hepatic veno-occlusive disease Budd–Chiari syndrome May–Thurner syndrome Portal vein thrombosis Renal vein thrombosis upper limb / torso Mondor's disease Paget–Schroetter disease head Cerebral venous sinus thrombosis Post-thrombotic syndrome Varicose veins Gastric varices Portacaval anastomosis Caput medusae Esophageal varices Hemorrhoid Varicocele Other Chronic venous insufficiency Chronic cerebrospinal venous insufficiency Superior vena cava syndrome Inferior vena cava syndrome Venous ulcer Arteries or veins Angiopathy Macroangiopathy Microangiopathy Embolism Pulmonary embolism Cholesterol embolism Paradoxical embolism Thrombosis Vasculitis Blood pressure Hypertension Hypertensive heart disease Hypertensive emergency Hypertensive nephropathy Essential hypertension Secondary hypertension Renovascular hypertension Benign hypertension Pulmonary hypertension Systolic hypertension White coat hypertension Hypotension Orthostatic hypotension
A number sign (#) is used with this entry because of evidence that pulmonary venoocclusive disease-1 (PVOD1) is caused by heterozygous mutation in the BMPR2 gene (600799) on chromosome 2q33. Description Pulmonary venoocclusive disease primarily affects the postcapillary venous pulmonary vessels and may involve significant pulmonary capillary dilation and/or proliferation. PVOD is an uncommon cause of pulmonary artery hypertension (PPH; see 178600), a severe condition characterized by elevated pulmonary artery pressure leading to right heart failure and death. PVOD accounts for 5 to 10% of 'idiopathic' PPH and has an estimated incidence of 0.1 to 0.2 cases per million. The pathologic hallmark of PVOD is the extensive and diffuse occlusion of pulmonary veins by fibrous tissue, with intimal thickening present in venules and small veins in lobular septa and, rarely, larger veins.
Moreover, the brain anomalies in the second patient, as indicated by CT and MRI scans, were predominantly in the basal ganglia and consistent with Leigh syndrome. The second patient manifested no dysmorphic facial features and no congenital heart disease and was still living at the time of report. ... Brain imaging showed abnormal signals in the globi pallidi reminiscent of Leigh syndrome. Laboratory studies showed increased lactate; 1 patient had increased hydroxy-C4-carnitine. ... Serial brain imaging showed a progressive reduction in brain volume, enlarged ventricles, and increasing signal abnormalities of the basal ganglia, reminiscent of Leigh syndrome. He never learned to sit independently and never acquired language. ... Using whole-exome sequencing, Stiles et al. (2015) identified a homozygous missense mutation in the HIBCH gene (R66W; 610690.0005) in 2 sibs with HIBCHD with features of Leigh syndrome. Sanger sequencing confirmed that both affected sibs were homozygous for this variant and that each parent was heterozygous, thus confirming recessive inheritance through the pedigree. INHERITANCE - Autosomal recessive HEAD & NECK Head - Head titubations (1 patient) Face - Dysmorphic facial features (in some patients) Eyes - Nystagmus - Strabismus - Epicanthal folds CARDIOVASCULAR Heart - Tetralogy of Fallot (1 patient) ABDOMEN Gastrointestinal - Poor feeding - Persistent vomiting MUSCLE, SOFT TISSUES - Hypotonia - Secondarily decreased activities of mitochondrial respiratory enzymes (in some patients) NEUROLOGIC Central Nervous System - Delayed psychomotor development - Developmental regression - Seizures - Myoclonus - Dystonia - Ataxia - Dysmetria - Abnormal movements - T2-weighted hyperintensities in the basal ganglia consistent with Leigh syndrome LABORATORY ABNORMALITIES - Increased lactate - Increased hydroxy-C4-carnitine - Urinary excretion of cysteine and cysteamine conjugates of methacrylic acid MISCELLANEOUS - Onset in infancy - Variable features MOLECULAR BASIS - Caused by mutation in the 3-@hydroxyisobutyryl-CoA hydrolase gene (HIBCH, 610690.0001 ) ▲ Close
To date, it has been described in four boys. The syndrome is caused by mutations affecting the two alleles of the HIBCH gene, encoding 3-hydroxyisobutyryl-CoA hydrolase.
HIBCH deficiency can cause signs and symptoms similar to another disease, called Leigh syndrome . Diagnosis is aided by blood tests which show high levels of lactic acid, and imaging studies which show changes in the "globi pallidi" structure of the brain.
The diagnosis is made by sequencing the mutated gene. [ citation needed ] Differential diagnosis [ edit ] Leigh syndrome Treatment [ edit ] There is currently no curative treatment for this condition.Supportive management is all that is currently available. [ citation needed ] History [ edit ] This condition was first described in 1982. [2] References [ edit ] ^ Yamada, Kenichiro; Naiki, Misako; Hoshino, Shin; Kitaura, Yasuyuki; Kondo, Yusuke; Nomura, Noriko; Kimura, Reiko; Fukushi, Daisuke; Yamada, Yasukazu; Shimozawa, Nobuyuki; Yamaguchi, Seiji; Shimomura, Yoshiharu; Miura, Kiyokuni; Wakamatsu, Nobuaki (2014).
For 2 years thereafter he displayed the nephrotic syndrome, followed in the next 2 years by uremia from which he died at age 39. ... The deposition of amyloid is characteristically interstitial rather than glomerular as seen in other forms of amyloidosis. The proband had the sicca syndrome. The details of their patient's family history were not given by Libbey and Talbert (1987). ... In 2 large American kindreds of Irish descent with nephrotic syndrome due to renal amyloidosis, Uemichi et al. (1993, 1994) identified a missense mutation in the FGA gene (E526V; 134820.0013). ... In mid-adult life, the patients developed slowly progressive chronic diarrhea with weight loss and sicca syndrome. One had sensorimotor axonal polyneuropathy and orthostatic hypotension and 2 had severe autonomic neuropathy. ... GU - Nephropathy with hematuria - Nephrotic syndrome - Uremia Neuro - Nonneuropathic Lab - Generalized amyloid deposition - Proteinuria - Hematuria Skin - Pitting edema - Petechial skin rash Endocrine - Hypertension Inheritance - Autosomal dominant Misc - Chronic weakness GI - Hepatomegaly - Cholestasis - Splenomegaly ▲ Close
A group of rare renal diseases, characterized by amyloid fibril deposition of apolipoprotein A-I or A-II (AApoAI or AApoAII amyloidosis), lysozyme (ALys amyloidosis) or fibrinogen A-alpha chain (AFib amyloidosis) in one or several organs. Renal involvement leading to chronic renal disease and renal failure is a common sign. Additional manifestations depend on the organ involved and the type of amyloid fibrils deposited.
In general, band heterotopia, also known as double cortex syndrome, [6] are seen exclusively in women; men with a mutation of the related gene (called XLIS or DCX ) usually die in utero or have a much more severe brain anomaly. Symptoms in affected women vary from normal to severe developmental delay or mental retardation; the severity of the syndrome is related to the thickness of the band of arrested neurons. Nearly all affected patients that come to medical attention have epilepsy , with partial complex and atypical absence epilepsy being the most common syndromes. Some of the more severely affected patients develop drop attacks. ... Subcortical band heterotopia, also known as “double cortex” syndrome, refers to a band of subcortical heterotopia neurons, located midway between the ventricles and the cerebral cortex. ... Heterotopia are most commonly isolated anomalies, but may be part of a number of syndromes, including chromosomal abnormalities and fetal exposure to toxins (including alcohol).
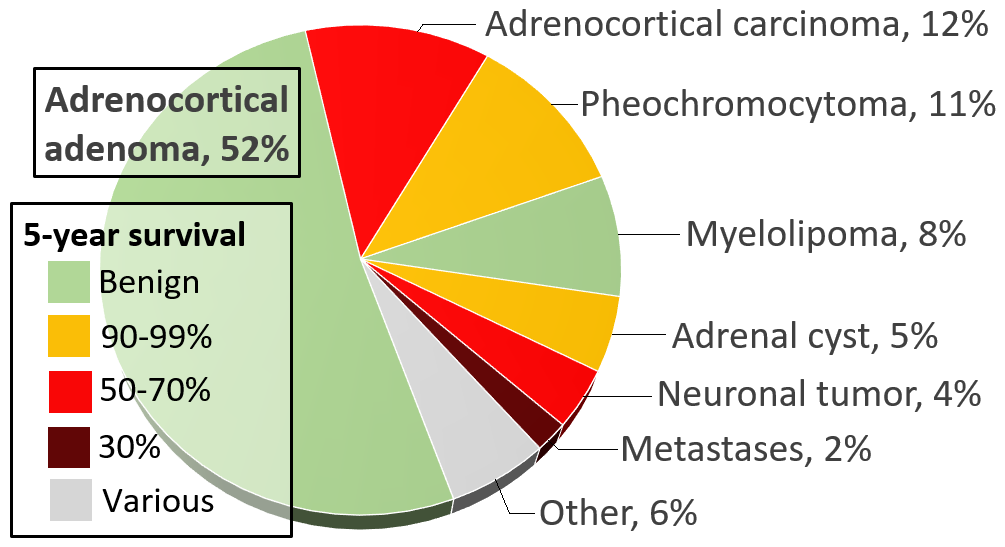
Second, a minority (about 15%) of adrenocortical adenomas are "functional", meaning that they produce glucocorticoids , mineralcorticoids , and/or sex steroids , resulting in endocrine disorders such as Cushing's syndrome , Conn's syndrome (hyperaldosteronism), virilization of females, or feminization of males. ... Pheochromocytomas occur in patients of all ages, and may be sporadic, or associated with a hereditary cancer syndrome , such as multiple endocrine neoplasia (MEN) types IIA and IIB, neurofibromatosis type I, or von Hippel-Lindau syndrome . ... Tumors under 3 cm are generally considered benign and are only treated if there are grounds for a diagnosis of Cushing's syndrome or pheochromocytoma . [7] Radiodensity gives a clue in estimating malignancy risk, wherein a tumor with 10 Hounsfield units or less on an unenhanced CT is probably a lipid-rich adenoma. [8] Hormonal evaluation includes: [9] 1-mg overnight dexamethasone suppression test 24-hour urinary specimen for measurement of fractionated metanephrines and catecholamines Blood plasma aldosterone concentration and plasma renin activity, if hypertension is present On CT scan, benign adenomas typically are of low radiographic density (due to fat content) and show rapid washout of contrast medium (50% or more of the contrast medium washes out at 10 minutes).
This variant has been described in cases of persistent aura without cerebral infarction . [10] See also [ edit ] Focal seizure – Epilepsy syndrome characterised by seizures preceded by isolated disturbances of a cerebral function Hallucination – Perception in the absence of external stimulation that has the qualities of real perception Persistent aura without infarction Synesthesia – Neurological condition involving the crossing of senses CADASIL Retinal migraine Photopsia References [ edit ] ^ Sudden Onset Panic: Epileptic Aura or Panic Disorder? ... External links [ edit ] Classification D ICD - 10 : G43.1 ICD - 9-CM : 346.0 Epilepsy.com information MAGNUM, the National Migraine Association Epilepsy Action information Migraines with 'aura' raise women's stroke risk v t e Seizures and epilepsy Basics Seizure types Aura (warning sign) Postictal state Epileptogenesis Neonatal seizure Epilepsy in children Management Anticonvulsants Investigations Electroencephalography Epileptologist Personal issues Epilepsy and driving Epilepsy and employment Seizure types Focal Seizures Simple partial Complex partial Gelastic seizure Epilepsy Temporal lobe epilepsy Frontal lobe epilepsy Rolandic epilepsy Nocturnal epilepsy Panayiotopoulos syndrome Vertiginous epilepsy Generalised Tonic–clonic Absence seizure Atonic seizure Automatism Benign familial neonatal seizures Lennox–Gastaut syndrome Myoclonic astatic epilepsy Epileptic spasms Status epilepticus Epilepsia partialis continua Complex partial status epilepticus Myoclonic epilepsy Progressive myoclonus epilepsy Dentatorubral–pallidoluysian atrophy Unverricht–Lundborg disease MERRF syndrome Lafora disease Juvenile myoclonic epilepsy Non-epileptic seizure Febrile seizure Psychogenic non-epileptic seizure Related disorders Sudden unexpected death in epilepsy Todd's paresis Landau–Kleffner syndrome Epilepsy in animals Organizations Citizens United for Research in Epilepsy (US) Epilepsy Action (UK) Epilepsy Action Australia Epilepsy Foundation (US) Epilepsy Outlook (UK) Epilepsy Research UK Epilepsy Society (UK)
COG4-CDG is an extremely rare form of CDG syndrome (see this term) characterized clinically in the single reported case to date by seizures, some dysmorphic features, axial hyponia, slight peripheral hypertonia and hyperreflexia.
A number sign (#) is used with this entry because of evidence that congenital disorder of glycosylation type IIj (CDG IIj, CDG2J) is caused by compound heterozygous mutation in the COG4 gene (606976) on chromosome 16q22. For a general discussion of CDGs, see CDG1A (212065). Clinical Features Reynders et al. (2009) reported a Portuguese boy, born of unrelated parents, with CDG type II. He presented at age 4 months with fever, progressive irritability, and complex seizures after a vaccination. He had mild dysmorphic features, such as down-sloping frontal area and thick hair, as well as mild neurologic signs, including axial hypotonia, mild peripheral hypertonia, and hyperreflexia. Laboratory studies showed increased serum transaminases, alkaline phosphatase, and LDH cholesterol, but decreased platelet count and coagulation factors.
COG5-CDG is an extremely rare form of CDG syndrome (see this term) characterized clinically in the single reported case to date by moderate mental retardation with slow and inarticulate speech, truncal ataxia, and mild hypotonia.
COG5 -congenital disorder of glycosylation ( COG5 -CDG, formerly known as congenital disorder of glycosylation type IIi) is an inherited condition that causes neurological problems and other abnormalities. The pattern and severity of this disorder's signs and symptoms vary among affected individuals. Individuals with COG5 -CDG typically develop signs and symptoms of the condition during infancy. These individuals often have weak muscle tone (hypotonia) and delayed development. Other neurological features include moderate to severe intellectual disability, poor coordination, and difficulty walking.
A number sign (#) is used with this entry because of evidence that congenital disorder of glycosylation type IIi (CDG IIi, CDG2I) is caused by homozygous mutation in the COG5 gene (606821) on chromosome 7q22. For a general discussion of CDGs, see CDG1A (212065) and CDG2A (212066). Clinical Features Paesold-Burda et al. (2009) described a 14-year-old Iraqi girl, born to remotely consanguineous parents, who at age 8 years demonstrated moderate mental retardation with slow and inarticulate speech, truncal ataxia, and mild hypotonia. Brain MRI revealed pronounced atrophy of the cerebellum and brainstem. Speech and cognition improved over the next several years, but mild hypotonia and ataxia persisted.
COG1-CDG is an extremely rare form of CDG syndrome (see this term) characterized clinically in the few cases reported to date by variable signs including microcephaly, growth retardation, psychomotor retardation and facial dysmorphism.
They described the disorder as a cerebrocostomandibular (CCMS; 117650)-like syndrome. The first boy was born of consanguineous Greek Turkish parents and presented with microcephaly, smooth philtrum, thin upper lip, short neck, low-set posteriorly rotated ears, Pierre-Robin sequence, growth retardation, and bilateral maculopathy. ... Zeevaert et al. (2009) described 2 patients with a cerebrocostomandibular-like syndrome in whom they identified a homozygous splice site mutation in the COG1 gene (606973.0002).
There is increased risk for myelodysplastic syndrome and, rarely, acute myelogenous leukemia. ... There is increased risk for myelodysplastic syndrome and, rarely, acute myelogenous leukemia. ... In five additional persons with PN: Three had myelodysplastic syndrome [Pianigiani et al 2001, Colombo et al 2012, Walne et al 2016]; Two had acute myelogenous leukemia [Porter et al 1999, Walne et al 2010]. ... While there is clinical overlap between PN and dyskeratosis congenita (DC) and Rothmund-Thomson syndrome (RTS), PN is clinically distinguishable from these two disorders (Table 2). ... Bone marrow abnormalities (e.g., premyelodysplastic changes) should be followed and managed by the consulting hematologist/oncologist. Management of myelodysplastic syndrome and acute myelogenous leukemia is per routine.
A type of skin cancer called squamous cell carcinoma, a precancerous blood disorder known as myelodysplastic syndrome (MDS), and a blood cancer called acute myelogenous leukemia that often follows MDS have occurred in a few people with PN.
There are some similarities to Rothmund-Thomson syndrome (RTS; 268400); however, the skin lesions of RTS primarily occur in sun-exposed areas, and the patients usually show marked alopecia of the head and eyebrows. ... Mostefai et al. (2008) noted the phenotypic overlap with the group of the major hereditary poikiloderma disorders, including Rothmund-Thomson syndrome, dyskeratosis congenita (127550), and Kindler syndrome (173650). ... Molecular Genetics In 3 affected sibs from a highly consanguineous Italian family with poikiloderma and neutropenia, who were known to be negative for mutation in the Rothmund-Thomson syndrome (RTS; 268400)-associated gene RECQL4 (603780), Volpi et al. (2010) identified homozygosity for a splice site mutation in the C16ORF57 gene (613276.0001).
Poikiloderma with neutropenia is a rare, genetic hereditary poikiloderma disorder characterized by early-onset poikiloderma (which typically begins in the extremities, progresses centripetally and eventually involves the trunk, face and ears) associated with chronic neutropenia, recurrent infections, pachyonychia and palmoplantar keratoderma. Growth and/or develomental delay and hepato- and/or splenomegaly are additional reported features.
Nomenclature The term " GARS1 -associated axonal neuropathy" includes an axonal form of CMT type 2 and a similar group of clinical syndromes classified as distal hereditary motor neuropathy or distal spinal muscular atrophy (dSMA-V). ... The pattern of hand involvement before leg involvement distinguishes dHMN-V from other dHMN subtypes. Silver syndrome is associated with spasticity in the legs and amyotrophy in the hands. Caused by pathogenic variants in BSCL2 , Silver syndrome is part of the spectrum of the BSCL2 -related neurologic disorders. ... The clinical pattern of disease onset with hand weakness and atrophy rather than foot involvement and absent sensory deficits in the early stages of the illness should raise a suspicion of carpal tunnel syndrome, neurogenic thoracic outlet syndrome, or multifocal motor neuropathy: In the absence of family history, paresthesia, and pain, the clinical pattern of median nerve dysfunction at the wrist in individuals with carpal tunnel syndrome may be similar to that seen in the early stages of GARS1 -associated axonal neuropathy. Carpal tunnel syndrome is usually asymmetric and limited to median nerve.
Several affected infants progressed to a myelodysplastic syndrome / leukemia-like condition within the first months of life. ... Disorders to Consider in the Differential Diagnosis of CLPB Deficiency View in own window Discriminative Feature Disorder Gene(s) MOI Additional Hallmarks 1 of the Disorder 3-MGA-uria 2 TAZ defect (Barth syndrome) TAZ XL In affected males: growth delay in infancy, cardiomyopathy (left ventricular non-compaction), neutropenia, myopathy, typical facial features, hypocholesterolemia, & a cognitive phenotype OPA3 defect (Costeff syndrome; OPA3 -related 3-methylglutaconic aciduria) OPA3 AR In infants: optic atrophy & movement disorder (ataxia or extrapyramidal disorder) SERAC1 defect (MEGDEL syndrome) SERAC1 AR Neonatal hypoglycemia & liver failure 3 In 2nd yr of life: progressive SNHL & neurologic manifestations (truncal hypotonia, spasticity of the limbs, dystonia, severe ID/DD, Leigh syndrome-like findings on MRI) TMEM70 defect (OMIM 614052) TMEM70 AR To date no specific syndromic presentation Typically in neonates: hyperammonemia, lactic acidosis, muscular hypotonia, hypertrophic cardiomyopathy, psychomotor retardation In individuals surviving neonatal period: DD DNAJC19 defect (DCMA syndrome) (OMIM 610198) DNAJC5 AR Characteristic combination of childhood-onset dilated cardiomyopathy, non-progressive cerebellar ataxia, testicular dysgenesis, growth failure AUH defect (3-methylglutaconyl-CoA hydratase deficiency) (OMIM 250950) AUH AR Adult-onset progressive spasticity & dementia w/characteristic slowly developing radiologic picture of extensive leukoencephalopathy 4 Uniquely distinguished by ↑ urinary excretion of 3-hydroxyisovaleric acid (3-HIVA) AGK defect (Sengers syndrome) (OMIM 212350) AGK AR Characteristic combination of bilateral cataracts, hypertrophic cardiomyopathy, & no to mild ID Can be lethal in neonatal period but survivors to adulthood w/mild involvement are known Not otherwise specified 3-MGA-uria (former 3-MGCA 4) Unknown Normal 3-methylglutaconyl-CoA hydratase enzyme activity & no defect in TAZ , OPA3 , SERAC1 , TMEM70 , DNAJC5 , AUH , or AGK Congenital neutropenia & cyclic neutropenia ELANE -related neutropenia ELANE AD Isolated neutropenia; no involvement of the CNS or other organs G6PC3 deficiency G6PC3 AR Presence of cardiovascular and/or urogenital abnormalities X-linked congenital neutropenia (see WAS -Related Disorders) WAS XL Isolated neutropenia; no involvement of CNS or other organs GATA1 -related X-linked cytopenia GATA1 XL Typical presentation in affected males: bleeding disorder & anemia; neutropenia occurs later Shwachman-Diamond syndrome SBDS AR Intestinal malabsorption due to exocrine pancreatic dysfunction Hyperekplexia Hereditary hyperekplexia ARHGEF9 GLRA1 GLRB GPHN SLC6A5 AD AR XL Generalized stiffness immediately after birth normalizes in 1st yrs of life.