For a more complete phenotypic description and information on genetic heterogeneity of Meckel syndrome, see MKS1 (249000). Clinical Features Frank et al. (2008) reported a consanguineous family of Kosovar Albanian origin in which 2 male fetuses were found to have ultrasonographic features of Meckel syndrome. ... Mapping By genomewide analysis of a family with Meckel syndrome, Frank et al. (2008) found linkage to a 3.2-Mb region on chromosome 12q21.31-q21.33 (lod score of 4.32). Molecular Genetics Meckel Syndrome To identify new Meckel syndrome loci, Baala et al. (2007) performed a genomewide linkage scan in 8 families unlinked to known Meckel syndrome loci and found linkage to chromosome 12. The interval was narrowed to an 8-Mb region containing the CEP290 gene which, in view of the phenotypic overlap between Joubert syndrome (213300) and Meckel syndrome, and the finding of Baala et al. (2007) of allelism of these 2 phenotypes at the MKS3 locus (607361), was considered an excellent candidate gene. ... Haplotype analysis indicated a founder effect. Meckel-like Cerebrorenodigital Syndrome Baala et al. (2007) also identified CEP290 mutations in 4 families presenting a cerebrorenodigital syndrome, with a phenotype between that of Meckel syndrome and Joubert syndrome and thus representing the continuum of the clinical spectrum between these 2 disorders.
A number sign (#) is used with this entry because of evidence that Meckel syndrome type 11 (MKS11) is caused by homozygous mutation in the TMEM231 gene (614949) on chromosome 16q23. For a general phenotypic description and a discussion of genetic heterogeneity of Meckel syndrome, see MKS1 (249000). Clinical Features Shaheen et al. (2013) reported a consanguineous Arab family in which 2 pregnancies were lost because of Meckel syndrome, which was diagnosed prenatally in both cases; 1 was spontaneously aborted, whereas the other was terminated. ... Molecular Genetics In a patient, born of consanguineous Arab parents, with Meckel syndrome type 11, Shaheen et al. (2013) identified a homozygous mutation in the TMEM231 gene (614949.0003).
Franceschini et al. (2004) observed 2 male sibs with features suggestive of Meckel syndrome (see MKS; 249000), including occipital encephalocele, polycystic kidneys, and polydactyly, but who also had short, incurved distal long bones and triradiate acetabula. ... Franceschini et al. (2004) noted that although short and bowed limbs are seen in about 15% of MKS cases (Majewski et al., 1983), the severity, bilaterality, and absolute symmetry of lower limb malformations in both sibs and the association with triradiate acetabula suggested a distinct syndrome.
A number sign (#) is used with this entry because of evidence that Meckel syndrome-13 (MKS13) and Joubert syndrome-29 (JBTS29) are caused by homozygous or compound heterozygous mutation in the TMEM107 gene (616183) on chromosome 17p13. ... For discussion of genetic heterogeneity of Meckel syndrome, see MKS1 (249000). For discussion of genetic heterogeneity of Joubert syndrome, see JBTS1 (213300). ... Features included occipital encephalocele, polydactyly, polycystic kidneys, micrognathia, contractures, and perinatal lethality. Joubert Syndrome 29 Lambacher et al. (2016) reported a 22-year-old man from the Caribbean with Joubert syndrome.
A number sign (#) is used with this entry because of evidence that Meckel syndrome type 3 (MKS3) is caused by homozygous or compound heterozygous mutation in the TMEM67 gene (609884) on chromosome 8q22. Description Meckel syndrome is an autosomal recessive pre- or perinatal lethal malformation syndrome characterized by renal cystic dysplasia and variably associated features including developmental anomalies of the central nervous system (typically occipital encephalocele), hepatic ductal dysplasia and cysts, and postaxial polydactyly (summary by Smith et al., 2006). For a more complete phenotypic description and information on genetic heterogeneity of Meckel syndrome, see MKS1 (249000). Clinical Features Morgan et al. (2002) stated that comparison of the clinical features of MKS3-linked cases with reports of MKS1- and MKS2 (603194)-linked kindreds suggested that polydactyly, and possibly encephalocele, are less common in MKS3-linked families. ... Consugar et al. (2007) identified 7 novel pathogenic mutations in the TMEM67 gene (see, e.g., 609884.0011) in 5 of 17 families with a clinical diagnosis of Meckel syndrome. All 5 families were of European origin. ... Heterogeneity Shaheen et al. (2011) identified a consanguineous Arab family in whom the Meckel-Gruber syndrome phenotype mapped to the MKS3 locus but in whom no mutation in the TMEM67 gene was found.
A number sign (#) is used with this entry because of evidence that Meckel syndrome type 2 (MKS2) is caused by homozygous mutation in the TMEM216 gene (613277) on chromosome 11q12. Mutation in the TMEM216 gene also causes Joubert syndrome type 2 (JBTS2; 608091). Description Meckel syndrome is a rare autosomal recessive lethal condition characterized by an occipital meningoencephalocele, enlarged kidneys with multicystic dysplasia and fibrotic changes in the portal area of the liver and with ductal proliferation, and postaxial polydactyly. ... Molecular Genetics In 3 fetuses from 2 Tunisian families with Meckel syndrome-2, Valente et al. (2010) identified a homozygous 341T-G mutation in the TMEM216 gene (L114R; 613277.0003). ... Six affected fetuses of 3 Palestinian families with Meckel syndrome-2 had a different truncating mutation in the TMEM216 gene (613277.0004). ... A Turkish family had 2 affected patients: 1 with Joubert syndrome and a fetus with Meckel syndrome, indicating that the 2 clinical disorders are part of the same spectrum.
Description Meckel syndrome, also known as Meckel-Gruber syndrome, is a severe pleiotropic autosomal recessive developmental disorder caused by dysfunction of primary cilia during early embryogenesis. ... Meckel syndrome type 7 (MKS7) has Dandy-Walker malformation as a more consistent feature. ... The presence of these abnormalities in all 3 sibs in the absence of polydactyly and encephalocele suggested that this is a distinct syndrome, but its distinctness from the Meckel syndrome was by no means certain. ... Trisomy 13, Meckel syndrome, and Down syndrome (190685) explained 255 of the 338 syndromic polydactyly cases. ... The frequency of non-NTD-related anomalies was 22.9% (8/35) in encephalocele. The Meckel-Gruber syndrome was the most frequently identified specific syndrome.
Meckel syndrome (MKS) is a rare, lethal, genetic, multiple congenital anomaly disorder characterized by the triad of brain malformation (mainly occipital encephalocele), large polycystic kidneys, and polydactyly, as well as associated abnormalities that may include cleft lip/palate, cardiac and genital anomalies, central nervous system (CNS) malformations, liver fibrosis, and bone dysplasia. ... Large polycystic kidneys with cystic dysplasia are a constant feature of Meckel syndrome. Hepatic dysgenesis and liver fibrosis are frequent. ... Most of these genes are also responsible for Joubert syndrome, leading to the concept that MKS is the extreme lethal phenotype of Joubert syndrome. ... Differential diagnosis Differential diagnoses include trisomy 13, Bardet-Biedl syndrome, Hydrolethalus, and Smith-Lemli-Opitz Syndrome. ... Management and treatment No treatment is currently available for Meckel syndrome which has a constantly fatal outcome.
Meckel syndrome is a very severe disorder that is characterized by multiple cysts on the kidneys, protrusion of a portion of the brain through an opening in the skull (occipital encephalocele), and extra fingers or toes (polydactyly). ... Because of these serious health problems, most infants with Meckel syndrome do not survive for long after birth. Meckel syndrome is caused by mutations in one of eight genes, and it is inherited in an autosomal-recessive manner.
A number sign (#) is used with this entry because of evidence that Meckel syndrome-9 (MKS9) can be caused by compound heterozygous mutation in the B9D1 gene (614144) on chromosome 17p11.2. One such family has been reported. Description Meckel syndrome is a severe autosomal recessive ciliopathy classically defined by the triad of encephalocele, polydactyly, and renal and biliary ductal dysplasia (summary by Hopp et al., 2011). For a general phenotypic description and a discussion of genetic heterogeneity of Meckel syndrome, see MKS1 (249000). Clinical Features Hopp et al. (2011) reported a fetus with Meckel syndrome who was found by ultrasound at 13 weeks' gestation to have posterior encephalocele and abnormal posterior fossa, bilaterally enlarged multicystic dysplastic kidneys, and no bladder.
A number sign (#) is used with this entry because Meckel syndrome type 5 (MKS5) is caused by homozygous or compound heterozygous mutation in the RPGRIP1L gene (610937) on chromosome 16q12. For a general description of Meckel syndrome, see MKS1 (249000). See also Joubert syndrome-7 (JBTS7; 611560), an allelic disorder with a less severe phenotype. Clinical Features Delous et al. (2007) reported 3 fetuses with Meckel syndrome diagnosed at 15 to 16 weeks' gestation by ultrasound. ... Molecular Genetics In 3 fetuses with Meckel syndrome type 5, Delous et al. (2007) identified homozygous or compound heterozygous truncating mutations in the RPGRIP1L gene (610937.0005-610937.0007) that all resulted in complete loss of protein function. INHERITANCE - Autosomal recessive HEAD & NECK Eyes - Microphthalmia Mouth - Cleft lip - Cleft palate ABDOMEN Liver - Bile duct proliferation GENITOURINARY Kidneys - Cystic renal disease SKELETAL Limbs - Bowing of the long bones Hands - Postaxial polydactyly Feet - Postaxial polydactyly NEUROLOGIC Central Nervous System - Occipital encephalocele - Anencephaly MISCELLANEOUS - Prenatal or perinatal death - Genetic heterogeneity - See Joubert syndrome 7 ( 611560 ), an allelic disorder with a less severe phenotype MOLECULAR BASIS - Caused by mutation in the RPGRIP1-like gene (RPGRIP1L, 610937.0005 ). ▲ Close
A number sign (#) is used with this entry because of evidence that Meckel syndrome-8 (MKS8) is caused by homozygous mutation in the TCTN2 gene (613846) on chromosome 12q24.31. One such family has been reported. Description Meckel-Gruber syndrome is a severe autosomal recessive ciliopathy classically defined by the triad of encephalocele, polydactyly, and renal and biliary ductal dysplasia. ... For a general phenotypic description and a discussion of genetic heterogeneity of Meckel syndrome, see MKS1 (249000). Clinical Features Shaheen et al. (2011) described a multiplex consanguineous Saudi family in which 5 individuals had Meckel-Gruber syndrome defined as the presence of encephalocele and any of the following: biliary ductal dysplasia, renal dysplasia, or polydactyly. ... Molecular Genetics In affected members of a consanguineous Arab family with Meckel-Gruber syndrome, Shaheen et al. (2011) identified homozygosity for a splice site mutation in the TCTN2 gene (613846.0001) that completely abolished normal splicing and created 2 aberrant transcripts.
A number sign (#) is used with this entry because Meckel syndrome type 6 (MKS6) is caused by homozygous mutation in the CC2D2A gene (612013) on chromosome 4p15. For a general phenotypic description and a discussion of genetic heterogeneity of Meckel syndrome, see MKS1 (249000). Clinical Features Tallila et al. (2008) reported the clinical features of probands from 11 Finnish families with Meckel syndrome. ... Mapping By genomewide SNP scan of 10 unrelated Finnish fetuses with Meckel syndrome, Tallila et al. (2008) found homozygosity in 6 for a shared overlapping region on chromosome 4p15. ... Molecular Genetics Tallila et al. (2008) selected the CC2D2A gene as a candidate for Meckel syndrome on chromosome 4p15 by searching a ciliary proteome database. ... Further analysis of 575 healthy controls indicated that the carrier frequency of the CC2D2A mutation was 0.5% in the Finnish population. In a patient with Meckel syndrome who was compound heterozygous for mutations in CC2D2A, Kim et al. (2010) identified heterozygosity for a missense mutation in the C2ORF86 gene (613580.0003).
Pilotto syndrome Other names Cleft lip and palate, congenital heart disease, scoliosis, short stature, and mental retardation [1] Pilotto syndrome is a rare syndrome which affects the face , heart , and back . The syndrome can cause a cleft lip and palate , scoliosis , and mental retardation . The Office of Rare Diseases and National Institutes of Health have classified this syndrome as affecting less than 200,000 people in the United States . [2] References [ edit ] ^ "Pilotto syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . Retrieved 17 March 2019 . ^ "Pilotto syndrome" . Health Grades Inc. Archived from the original on 9 February 2010 .
Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( January 2021 ) Retained antrum syndrome Other names Retained gastric antrum syndrome Retained antrum syndrome or retained gastric antrum syndrome is one of the rare postgastrectomy syndromes . It happens after Billroth II surgery and the mechanism involved is the inadequate removal of the distal antrum and pylorus during resection of the antrum and gastrojejunostomy . [1] [2] Lack of the usual acid-secreting gastric glands makes the antral segment continually exposed to the alkaline environment of the duodenum, which causes it to secrete excessive acid and be prone to form ulcers . [2] References [ edit ] ^ "retained antrum syndrome" . ^ a b "retained gastric antrum syndrome" .
Nephrotic syndrome-deafness-pretibial epidermolysis bullosa syndrome is a rare, genetic, renal disease characterized by hereditary nephritis leading to nephrotic syndrome and end-stage renal failure associated with sensorineural hearing loss and pretibial skin blistering followed by atrophy.
A number sign (#) is used with this entry because of evidence that this syndrome is caused by mutation in the CD151 gene (602243). ... Association of deafness and nephropathy suggested Alport syndrome, which is usually inherited as an X-linked disorder (301050) and caused by mutation in the COL4A5 gene (303630). Autosomal recessive Alport syndrome (203780) is caused by mutation in the COL4A3 (120070) or COL4A4 (120131) genes.
This disorder causes a progressive loss of motor skills and language. Rett syndrome primarily affects females. Most babies with Rett syndrome seem to develop as expected for the first six months of life. ... Rett syndrome can also cause seizures and intellectual disabilities. ... Although there's no cure for Rett syndrome, potential treatments are being studied. ... In a very small number of cases, inherited factors — for instance, having close family members with Rett syndrome — may play a role. Complications Complications of Rett syndrome include: Sleep problems that cause significant sleep disruption to the person with Rett syndrome and family members. ... Evaluating other causes for the symptoms Because Rett syndrome is rare, your child may have certain tests to determine whether other conditions are causing some of the same symptoms as Rett syndrome.
Clinical description Classic or typical Rett syndrome (RTT) primarily affects girls and is characterized by apparently normal psychomotor development during the first 6-18 months of life followed by developmental stagnation with rapid regression in language and motor abilities, and subsequent long-term plateauing of skills. ... Differential diagnosis Differential diagnosis includes autism spectrum disorder; Angelman syndrome in which early development before 6 months is abnormal; CDKL5 deficient disorders in which early onset seizures are distinctive from RTT; FOXG1 syndrome in which congenital microcephaly and corpus callosum abnormalities are not usually found in RTT.
(Rettsearch Consortium) (2010). "Rett syndrome: Revised diagnostic criteria and nomenclature" . ... "Early motor disturbances in Rett syndrome and its pathophysiological importance". ... ISBN 978-1-58562-444-7 . ^ a b "Rett syndrome Tests and diagnosis" . Mayo Clinic . Archived from the original on 30 October 2017. ^ a b c d e f "About Rett syndrome - Rett Syndrome Diagnosis" . www.rettsyndrome.org . International Rett Syndrome Foundation. Archived from the original on 29 October 2017 .
Rett syndrome is a progressive, neuro-developmental condition that primarily affects girls. ... Some people have an atypical form of Rett syndrome that may be more mild or more severe. Classic Rett syndrome is most commonly caused by mutations in the MECP2 gene and is usually inherited in an X-linked dominant manner.
During the neonatal period and infancy it may also include aplasia cutis congenita, herpes simplex infection, epidermolytic ichthyosis, bullous impetigo, staphylococcal scalded skin syndrome, linear IgA bullous dermatosis, bullous pemphigoid, neonatal pemphigus and pemphigoid gestationis (see these terms).
Dystrophic epidermolysis bullosa (DEB) is one of the major forms of epidermolysis bullosa . The signs and symptoms can vary widely among affected people. In mild cases, blistering may primarily affect the hands, feet, knees, and elbows. Severe cases often involve widespread blistering that can lead to vision loss, disfigurement, and other serious medical problems. DEB is caused by changes (mutations) in the COL7A1 gene and may be inherited in an autosomal dominant or autosomal recessive manner depending on the subtype. New blisters should be lanced, drained, and protected. Some patients need nutritional support, supplements, occupational therapy and/or surgery depending on the associated features of the disease.
Differential Diagnosis The four major types of epidermolysis bullosa (EB) syndrome, caused by pathogenic variants in 20 different genes, are EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB), and Kindler syndrome (see Table 4). ... Epidermolysis Bullosa Types View in own window Clinical Features 1 EB Type EB Subtype Gene MOI Skin Biopsy Findings (Level of Cleavage) Extent of Blistering Presence of Oral/ Other Mucous Membrane Lesions Presence/Extent of Scarring Other (Associated Gene) 1 Mild to severe depending on gene &/or variant Mucous membrane involvement in severe forms Not a consistent feature Scarring reported in KLHL24 -related EB Severe keratoderma Dilated cardiomyopathy &/or woolly hair ( DSP ) Ectodermal dysplasia ( PKP1 ) Right ventricular cardiomyopathy ( JUP ) Kindler-like phenotype w/neuropathy ( CD151 ) Muscular dystrophy &/or pyloric atresia ( PLEC ) Hereditary sensory & autonomic neuropathy Type VI ( DST ) Alopecia & cardiomyopathy ( KLHL24 ) EBS 3 EBS suprabasal TGM5 AR In all EBS: a split above dermal-epidermal junction at ultrastructural level DSP PKP1 JUP EBS basal KRT5 AD (AR 2 ) KRT14 CD151 EXPH5 AR PLEC DST KLHL24 Moderate to severe depending on type of pathogenic variant Tooth enamel involvement (amelogenesis imperfecta) Oral/eye/throat lesions (JEB-LOC syndrome 4 ) Granulation tissue formed on skin around oral & nasal cavities, fingers, & toes, & internally around upper airway & nails suggests JEB-GS. Hoarseness Prone to sepsis Alopecia ( COL17A1 especially) Urogenital anomalies ( ITGB4 ) Congenital interstitial lung disease, nephrotic syndrome ( ITGA3 ) JEB JEB severe generalized LAMA3 AR In all JEB: a split at level of lamina lucida, either: W/in lamina lucida; or Occasionally, just above lamina lucida at hemidesmosomes (seen w/pathogenic variants in COL17A1 , ITGB4 , & ITGA6 ) LAMB3 LAMC2 JEB generalized & localized LAMA3 LAMB3 LAMC2 COL17A1 ITGB4 JEB late onset COL17A1 JEB w/pyloric atresia ITGB4 ITGA6 JEB w/respiratory & renal involvement ITGA3 JEB-LOC syndrome 4 LAMA3A Extensive; predominantly on hands & feet Esophageal strictures in RDEB Pseudosyndactyly (mitten deformities) caused by scarring of hands & feet in older children & adults in RDEB Corneal erosions Dystrophic or nail loss DEB RDEB severe generalized COL7A1 AR Forms below the basement membrane (in superficial dermis) RDEB generalized & localized DDEB (all subtypes) AD Fragile thin skin w/acral blistering Mucous membrane involvement Esophageal strictures in adulthood Skin atrophy w/tissue paper appearance Photosensitivity Progressive poikiloderma Diffuse cutaneous atrophy Telangiectasia Depigmentation Kindler syndrome NA FERMT1 AR Multiple cleavage planes; specifically epidermal, lamina lucida, & sublamina densa (multiple cleavage planes are unique to mechanobullous disorders) Partially adapted from Fine et al [2014] AD = autosomal dominant; AR = autosomal recessive; DDEB = dominant dystrophic epidermolysis bullosa; EB = epidermolysis bullosa; EBS = epidermolysis bullosa simplex; JEB = junctional epidermolysis bullosa; JEB-GS = generalized severe junctional epidermolysis bullosa; MOI = mode of inheritance; RDEB = recessive dystrophic epidermolysis bullosa 1. ... EBS is divided into suprabasal and basal EBS defined by the level of the cleavage. 4. Laryngoonychocutaneous (LOC) syndrome, or JEB-LOC (OMIM 245660), is described in Punjabi Indians.
This type of syndrome can result from a unilateral lesion in the brainstem affecting both upper motor neurons and lower motor neurons . ... If the symptoms can be attributed to another disease or disorder, then a definitive diagnosis is difficult to make. [6] : 778 Diagnosis of Weber's syndrome [ edit ] Weber's syndrome is the only form of alternating hemiplegia that is somewhat easy to diagnose beyond the general criteria. Although Weber's syndrome is rare, a child born with the disorder typically has a port-wine stain on the face around the eye. While the port-wine stain does not necessarily mean the child has Weber's syndrome, if the port-wine stain involves the ophthalmic division of the trigeminal nerve than the likelihood of it being weber's syndrome greatly increases. ... External links [ edit ] Classification D OMIM : 602481 DiseasesDB : 4693 alternatinghemiplegia at NINDS v t e Symptoms , signs and syndromes associated with lesions of the brain and brainstem Brainstem Medulla (CN 8, 9, 10, 12) Lateral medullary syndrome/Wallenberg PICA Medial medullary syndrome/Dejerine ASA Pons (CN 5, 6, 7, 8) Upper dorsal pontine syndrome/Raymond-Céstan syndrome Lateral pontine syndrome ( AICA ) (lateral) Medial pontine syndrome / Millard–Gubler syndrome / Foville's syndrome ( basilar ) Locked-in syndrome Internuclear ophthalmoplegia One and a half syndrome Midbrain (CN 3, 4) Weber's syndrome ventral peduncle, PCA Benedikt syndrome ventral tegmentum, PCA Parinaud's syndrome dorsal, tumor Claude's syndrome Other Alternating hemiplegia Cerebellum Latearl Dysmetria Dysdiadochokinesia Intention tremor ) Medial Cerebellar ataxia Basal ganglia Chorea Dystonia Parkinson's disease Cortex ACA syndrome MCA syndrome PCA syndrome Frontal lobe Expressive aphasia Abulia Parietal lobe Receptive aphasia Hemispatial neglect Gerstmann syndrome Astereognosis Occipital lobe Bálint's syndrome Cortical blindness Pure alexia Temporal lobe Cortical deafness Prosopagnosia Thalamus Thalamic syndrome Other Upper motor neuron lesion Aphasia
1p36 deletion syndrome Other names Monosomy 1p36 A toddler showing facial symptoms of the syndrome. 1p36 deletion syndrome is a congenital genetic disorder characterized by moderate to severe intellectual disability , delayed growth, hypotonia , seizures, limited speech ability, malformations, hearing and vision impairment, and distinct facial features . ... It is one of the most common deletion syndromes. It is estimated that the syndrome occurs in one in every 5,000 to 10,000 births. ... Feeding difficulties can be managed with specialized assistive devices or with a gastrostomy (feeding) tube . [2] Epidemiology [ edit ] 1p36 deletion syndrome is the most common terminal deletion syndrome in humans. [5] It occurs in between 1 in 5000 and 1 in 10000 live births. [3] References [ edit ] ^ "Chromosome 1, 1p36 deletion syndrome" . ... (eds.), "1p36 Deletion Syndrome" , GeneReviews® , University of Washington, Seattle, PMID 20301370 , retrieved 2019-02-20 ^ a b "OMIM Entry - # 607872 - CHROMOSOME 1p36 DELETION SYNDROME" . www.omim.org . Retrieved 19 September 2018 . ^ "Chromosome 1p36 deletion syndrome" . Genetics Testing Reference .
1p36 deletion syndrome is a disorder that typically causes severe intellectual disability. ... Some have abnormalities of the skeleton, heart, gastrointestinal system, kidneys, or genitalia. Frequency 1p36 deletion syndrome is believed to affect between 1 in 5,000 and 1 in 10,000 newborns. ... The signs and symptoms of 1p36 deletion syndrome are probably related to the loss of multiple genes in this region. ... Learn more about the chromosome associated with 1p36 deletion syndrome chromosome 1 Inheritance Pattern Most cases of 1p36 deletion syndrome are not inherited. ... About 20 percent of people with 1p36 deletion syndrome inherit the chromosome with a deleted segment from an unaffected parent.
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome caused by haploinsufficiency of a number of genes. Description The constitutional deletion of chromosome 1p36 results in a syndrome with multiple congenital anomalies and mental retardation (Shapira et al., 1997). Monosomy 1p36 is the most common terminal deletion syndrome in humans, occurring in 1 in 5,000 births (Shaffer and Lupski, 2000; Heilstedt et al., 2003). ... Tan et al. (2005) reported a 16-year-old boy with features of Cantu syndrome (239850) who was found to have a distal 1p36 deletion. ... There was an approximately 6.9- to 7.1-Mb deletion from chromosome 1p36.33 to 1p36.23, containing the critical region for 1p36 deletion syndrome, as well as an approximately 5.6- to 6.0-Mb duplication from 21q22.3 to 21qter, distal to the Down syndrome (190685) critical region.
1p36 deletion syndrome is a chromosomal anomaly characterized by distinctive facial dysmorphic features, hypotonia, developmental delay, intellectual disability, seizures, heart defects, hearing impairment and prenatal onset growth deficiency. Epidemiology 1p36 deletion syndrome is considered one of the most common chromosome deletion syndromes, with an incidence of 1/ 5,000 to 1 /10,000 live births occurring equally in both genders, across all ethnicities. ... Differential diagnosis Differential diagnosis includes Rett syndrome, Angelman syndrome and Prader-Willi syndrome (see these terms). ... Prognosis The severity of the 1p36 deletion syndrome varies between affected individuals. ... Patients will remain dependent of others for most activities of daily living and will require medical support throughout life. Individuals with 1p36 deletion syndrome survive well into adult life.
Description Neurodevelopmental disorder with or without anomalies of the brain, eye, or heart is an autosomal dominant syndrome characterized by onset in infancy of developmental delay, intellectual disability, and behavioral disorders, such as autism spectrum disorders. ... The phenotype is reminiscent of that observed in patients with 1p36 deletion syndrome (607872); RERE is located in the proximal 1p36 critical region (summary by Fregeau et al., 2016).
1p36 deletion syndrome is a chromosome disorder that typically causes severe intellectual disability. ... Other features include a small head that is unusually short and wide; vision and hearing problems; abnormalities of the skeleton, heart, gastrointestinal system, kidneys, or genitalia; and distinctive facial features. 1p36 deletion syndrome is caused by a deletion of genetic material from a specific region in the short (p) arm of chromosome 1 . Most cases are not inherited; only about 20% of the cases of people with 1p36 deletion syndrome inherit the chromosome with a deleted segment from an unaffected parent. In these cases, the parent carries a balanced translocation, in which no genetic material is gained or lost. There is no cure for this syndrome. Treatment depends on the symptoms, and may include rehabilitation/educational programs, antiepileptic medication, and standard treatment for heart, kidney, eye, hearing or bone problems.
Overview Toxic shock syndrome is a rare, life-threatening complication of certain types of bacterial infections. ... Toxic shock syndrome can affect anyone, including men, children and postmenopausal women. ... Causes Most commonly, Staphylococcus aureus (staph) bacteria cause toxic shock syndrome. The syndrome can also be caused by group A streptococcus (strep) bacteria. Risk factors Toxic shock syndrome can affect anyone. About half the cases of toxic shock syndrome associated with staphylococci bacteria occur in women of menstruating age; the rest occur in older women, men and children. ... Treatment If you develop toxic shock syndrome, you'll likely be hospitalized.
"Toxic shock syndrome: major advances in pathogenesis, but not treatment". ... S2CID 30680160 . ^ "Toxic shock syndrome (other than Streptococcal) (TSS): 2011 Case Definition" . ... "Clindamycin-induced suppression of toxic-shock syndrome-associated exotoxin production". ... "A New Generation Faces Toxic Shock Syndrome" . The Seattle Times . Knight Ridder Newspapers. first published as "Lingering Risk", San Jose Mercury News , 13 December 2004 ^ "Stayfree — FAQ About Toxic Shock Syndrome (TSS)" . 2006. ... PMC 2626872 . PMID 8903167 . "Toxic Shock Syndrome (TSS): The Facts" . Toxic Shock Syndrome information service . tssis.com.
Webbed toes occur most commonly in the following circumstances: [ citation needed ] Syndactyly or familial syndactyly Down syndrome It is also associated with a number of rare conditions, notably: Aarskog–Scott syndrome Acrocallosal syndrome Apert syndrome Bardet–Biedl syndrome Carpenter syndrome Cornelia de Lange syndrome Edwards syndrome Jackson–Weiss syndrome Fetal hydantoin syndrome Miller syndrome Pfeiffer syndrome Smith–Lemli–Opitz syndrome Timothy syndrome Ectodermal dysplasia Klippel–Feil syndrome Diagnosis [ edit ] This condition is normally discovered at birth. If other symptoms are present, a specific syndrome may be indicated. [ citation needed ] Diagnosis of a specific syndrome is based on family history, medical history, and a physical exam. ... External links [ edit ] Classification D ICD - 10 : Q70.3 ICD - 9-CM : 755.13 External resources MedlinePlus : 003289 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum
Aarskog-Scott syndrome is a genetic disorder that affects the development of many parts of the body. This condition mainly affects males, although females may have mild features of the syndrome. People with Aarskog-Scott syndrome often have distinctive facial features, such as widely spaced eyes (hypertelorism ), a small nose, a long area between the nose and mouth (philtrum ), and a widow's peak hairline . ... Most males with Aarskog-Scott syndrome have a shawl scrotum, in which the scrotum surrounds the penis instead of hanging below. ... The cause of Aarskog-Scott syndrome in other affected individuals is unknown. Learn more about the gene associated with Aarskog-Scott syndrome FGD1 Inheritance Pattern When caused by FGD1 gene mutations, Aarskog-Scott syndrome is inherited in an X-linked recessive pattern .
Contents 1 History 2 Signs and symptoms 3 Genetics 4 Treatment 5 References 6 External links History [ edit ] Mauriac syndrome was first described in 1930 by Pierre Mauriac . [1] It was described as a syndrome of growth failure and delayed puberty in children with poorly-controlled type I diabetes. ... These symptoms can be reversed with good glycemic control. [2] [3] [4] Genetics [ edit ] Abnormally high blood sugar levels are relatively common among patients with type I diabetes, but Mauriac syndrome is rare. This suggests that high blood sugar is not the only factor necessary to cause the syndrome. ... The child's father had type 1 diabetes. Neither parent had Mauriac syndrome. [1] The study suggests that both the mutant enzyme and an abnormally high blood glucose level were necessary to cause Mauriac syndrome. ... PMID 17360304 . ^ Kim MS, Quintos JB (August 2008). "Mauriac syndrome: growth failure and type 1 diabetes mellitus". ... PMID 18806715 . ^ Kocova, Mirjana; Milenkova, Liljana (4 July 2018). "Old syndrome–new approach: Mauriac syndrome treated with continuous insulin delivery" .
Metabolic syndrome Other names Dysmetabolic syndrome X A man with marked central obesity, a hallmark of metabolic syndrome. ... The cause of the syndrome is an area of ongoing medical research . ... "Prevalence and trends of metabolic syndrome in the adult U.S. population, 1999–2010" . ... "The obesity epidemic, metabolic syndrome and future prevention strategies". ... "Paleolithic nutrition for metabolic syndrome: systematic review and meta-analysis" .
When to see a doctor If you know you have at least one component of metabolic syndrome, ask your doctor whether you need testing for other components of the syndrome. Causes Metabolic syndrome is closely linked to overweight or obesity and inactivity. ... Risk factors The following factors increase your chances of having metabolic syndrome: Age. Your risk of metabolic syndrome increases with age. ... Other diseases. Your risk of metabolic syndrome is higher if you've ever had nonalcoholic fatty liver disease, polycystic ovary syndrome or sleep apnea. Complications Having metabolic syndrome can increase your risk of developing: Type 2 diabetes.
Dahlberg Borer Newcomer syndrome Other names Lymphedema hypoparathyroidism syndrome , Hypoparathyroidism lymphedema syndrome , Dahlberg syndrome . [1] Dahlberg Borer Newcomer syndrome is a rare autosomal X-linked recessive genetic condition characterized by a prolapse of the bicuspid valve , progressive kidney failure, congenital lymphedema , hypoparathyroidism , and very short end bones of fingers. [2] Contents 1 Treatments 2 References 3 Further reading 4 External links Treatments [ edit ] Treatment for this condition is based on its symptoms. These treatments may include manual lymphatic drainage , consumption of beta blockers or anticoagulants for the bicuspid valve prolapse and vitamin D or calcium carbonate tablets for the hypoparathyroidism. [1] References [ edit ] ^ a b "Lymphedema Hypoparathyroidism Syndrome" . Lymphedema People. 2008-05-29 . ... "Autosomal or X-linked recessive syndrome of congenital lymphedema, hypoparathyroidism, nephropathy, prolapsing mitral valve, and brachytelephalangy". ... Further reading [ edit ] Dahlberg Borer Newcomer syndrome at Orphanet External links [ edit ] Classification D ICD - 10 : Q87.8 OMIM : 247410 MeSH : C535769 External resources Orphanet : 1563
A rare multiple congenital anomalies/dysmorphic syndrome characterized by the association of congenital hypoparathyroidism, nephropathy, congenital lymphedema, mitral valve prolapse and brachytelephalangy.
Dahlberg et al. (1983) described 2 adult brothers with congenital lymphedema, hypoparathyroidism, nephropathy, mitral valve prolapse and brachytelephalangy. The older sib was found to have bilateral cataracts on routine examination at age 19 years. Swelling of his arms and legs, noted soon after his birth, increased after he began walking. Progressive renal failure necessitated renal transplantation at age 26 years. The brother had similar findings. Both have a broad nasal bridge and lateral displacement of the inner canthi.
Gordon syndrome Other names Camptodactyly-cleft palate-clubfoot syndrome Gordon syndrome is inherited in an autosomal dominant manner Gordon syndrome , or distal arthrogryposis type 3, is a rare genetic disorder characterized by cleft palate and congenital contractures of the hands and feet. ... Intelligence is not affected. [1] [2] Cause [ edit ] Gordon syndrome is a rare autosomal dominant disorder caused by mutation in PIEZO2 . [2] Epidemiology [ edit ] It affects males and females equally. Fewer than 50 cases have been reported worldwide. [1] References [ edit ] ^ a b "Gordon Syndrome" . NORD (National Organization for Rare Disorders) . Retrieved 15 April 2019 . ^ a b "Gordon syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . Retrieved 15 April 2019 . Gordon syndrome. Orphanet. February 2005; http://www.orpha.net/consor/cgi-bin/OC_Exp.php?
There are 2 syndromes with features overlapping those of DA3 that are also caused by heterozygous mutation in PIEZO2: distal arthrogryposis type 5 (DA5; 108145) and Marden-Walker syndrome (MWKS; 248700), which are distinguished by the presence of ocular abnormalities and mental retardation, respectively. ... Among the 6 affected, clubfoot occurred in 5, camptodactyly in 4, and cleft palate in 3. A useful list of camptodactyly syndromes was provided. Higgins et al. (1972) studied a father and 2 children with the same syndrome. ... Moldenhauer (1964) described 4 females of 3 generations of a family with a condition he called Nielson syndrome. The features were short stature, ptosis, cleft palate, camptodactyly, pterygium colli, and vertebral anomalies. ... They pointed to reduced penetrance and carrier females as a cardinal feature of the Gordon syndrome. Schrander-Stumpel et al. (1993) described an isolated case of arthrogryposis, ophthalmoplegia, and retinopathy in a Dutch family. ... Becker and Splitt (2001) reported a mother and 2 affected children with distal arthrogryposis and cleft palate and suggested a clinical overlap between Gordon syndrome and Aase-Smith syndrome (147800).
Gordon Syndrome is a rare, inherited disorder that affects movement in the joints of the upper and lower limbs, also known as distal arthrogryposis . ... The range and severity of symptoms may vary from person to person. Gordon syndrome is caused by genetic changes (mutations) in the PIEZO2 gene and can be inherited in an autosomal dominant pattern. Gordon syndrome is diagnosed by clinical examination and genetic testing. Treatment is directed at the symptoms and includes surgery and physical therapy to loosen the joints. People with Gordon syndrome often have reduced mobility, but Gordon syndrome doesn't usually result in other health problems.
Gordon syndrome, also known as distal arthrogryposis type 3, is an extremely rare multiple congenital malformation syndrome characterized by congenital contractures of hand and feet with variable degrees of severity of camptodactyly, clubfoot and, less frequently, cleft palate. Intelligence is normal but in some cases, additional abnormalities, such as short stature, kyphoscoliosis, ptosis, micrognathia, and cryptorchidism may also be present. Gordon syndrome, Marden-Walker syndrome and arthrogryposis with oculomotor limitation and electroretinal anomalies clinically and genetically overlap, and could represent variable expressions of the same condition.
Find sources: "Ho–Kaufman–Mcalister syndrome" – news · newspapers · books · scholar · JSTOR ( July 2014 ) ( Learn how and when to remove this template message ) Ho–Kaufman–Mcalister syndrome Other names Chen-Kung Ho–Kaufman–Mcalister syndrome Ho–Kaufman–Mcalister syndrome , is a rare congenital malformation syndrome where infants are born with a cleft palate , micrognathia , Wormian bones , congenital heart disease , dislocated hips , bowed fibulae , preaxial polydactyly of the feet, abnormal skin patterns, and most prominently, missing tibia . The etiology is unknown. Ho–Kaufman–Mcalister syndrome is named after Chen-Kung Ho , R.L. ... Mcalister who first described the syndrome in 1975 at Washington University in St. ... References [ edit ] Ho–Kaufman–Mcalister Syndrome Summary at PubMed Ho CK, Kaufman RL, Mcalister WH (1975).
Extracutaneous manifestations like testicular maldescent and/or corneal opacities are possible for rare cases, if the ichthyosis is part of a disease syndrome (syndromic RXLI; see this term). ... STS deficiency leads to increased amounts of CSO4 that inhibit epidermal serine proteases, which in turn results in decreased desquamation of corneocytes with retention hyperkeratosis. There are some much more rare syndromic RXLI cases that are due to contiguous gene deletion affecting neighbouring genes of the STS gene. This can be observed in Kallman syndrome, hypergonadotropic hypogonadism, ocular albinism type 1 (see these terms), or hypertrophic pyloric stenosis. ... Differential diagnosis Differential diagnosis includes ichthyosis vulgaris, autosomal recessive congenital ichthyosis (ARCI), namely lamellar ichthyosis, syndromic RLXI, or multiple sulfatase deficiency (see these terms). ... Therefore, RXLI may be detected in utero , when maternal estriol levels are measured for prenatal screening for Down syndrome and other disorders. Genetic counseling XLRI is transmitted as an X-linked recessive trait: it affects males and is inherited through female carriers.
Keppen–Lubinsky syndrome Other names Generalized lipodystrophy-progeroid features-severe intellectual disability syndrome This condition is inherited in an autosomal dominant manner Keppen–Lubinsky syndrome is an extremely rare congenital disorder . The minimal clinical criteria for the Keppen–Lubinsky syndrome are as follows: normal growth parameters at birth, postnatal growth failure, peculiar face with an aged appearance (large prominent eyes, a narrow nasal bridge, a tented upper lip, a high palate, an open mouth), skin tightly adherent to facial bones, generalized lipodystrophy , microcephaly, and development delay. [1] [2] [3] Keppen-Lubinsky syndrome is caused by mutation in the inwardly rectifying K+ channels encoded by KCNJ6 gene. [4] References [ edit ] ^ Gorlin, Robert; Cohen, M. Michael; Hennekam, Raoul (2001). "Syndromes of the head and neck" . Keppen–Lubinsky syndrome (4th ed.). ... ISBN 9780199747726 . ^ De Brasi, D; Brunetti-Pierri, N; Di Micco, P; Andria, G; Sebastio, G (2003). "New syndrome with generalized lipodystrophy and a distinctive facial appearance: confirmation of Keppen-Lubinski syndrome?". ... S2CID 38839849 . ^ Basel-Vanagaite, Lina; Shaffer, Lisa; Chitayat, David (2009). "Keppen-Lubinsky syndrome: Expanding the phenotype". American Journal of Medical Genetics . 149A (8): 1827–9. doi : 10.1002/ajmg.a.32975 .
A number sign (#) is used with this entry because of evidence that Keppen-Lubinsky syndrome (KPLBS) is caused by heterozygous mutation in the KCNJ6 gene (600877) on chromosome 21q22. Description Keppen-Lubinsky syndrome is a very rare disorder characterized by severely delayed psychomotor development, hypertonia, hyperreflexia, generalized lipodystrophy giving an aged appearance, and distinctive dysmorphic features, including microcephaly, prominent eyes, narrow nasal bridge, and open mouth (summary by Masotti et al., 2015). ... All growth parameters were low; brain MRI was normal. Beare-Stevenson cutis gyrata syndrome (BSTVS; 123790) was excluded. De Brasi et al. (2003) reported an 8-year-old boy with generalized lipodystrophy, peculiar aged facial appearance with tight, thin skin overlying the facial bones and small pinched nose, and almost complete lack of psychomotor development. They noted the similarities to the patient reported by Gorlin et al. (2001), and suggested that this is a novel progeroid syndrome characterized by normal growth parameters at birth, very poor postnatal growth, failure to thrive, and developmental delay. Basel-Vanagaite et al. (2009) reported a third boy with features consistent with Keppen-Lubinsky syndrome. He was born of unrelated Jewish parents of Yemenite origin and had a normal birth weight.
A rare, genetic, primary lipodystrophy syndrome characterized by severe developmental delay and intellectual disability, hypertonia, hyperreflexia, microcephaly, tightly adherent skin, an aged appearance, severe generalized lipodystrophy, and distinct facial dysmorphism which includes large prominent eyes, narrow nasal bridge, tented upper lip vermilion, an open mouth, and high-arched palate.
Woodhouse–Sakati syndrome Woodhouse–Sakati syndrome has an autosomal recessive pattern of inheritance. Woodhouse–Sakati syndrome , [1] is a rare autosomal recessive [2] multisystem disorder which causes malformations throughout the body, and deficiencies affecting the endocrine system . [3] Contents 1 Presentation 2 Genetics 3 Diagnosis 4 Treatment 5 References 6 External links Presentation [ edit ] The syndrome is characterized by alopecia , hypogonadism , hypothyroidism , hearing loss , intellectual disability and diabetes mellitus . ... You can help by adding to it . ( August 2017 ) References [ edit ] ^ a b Online Mendelian Inheritance in Man (OMIM): 241080 ^ a b c Medica I, Sepcic J, Peterlin B (2007). "Woodhouse–Sakati syndrome: case report and symptoms review". ... PMID 17710875 . ^ Woodhouse NJ, Sakati NA (1983). "A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities" . ... PMID 6876115 . ^ Online Mendelian Inheritance in Man (OMIM): 612515 External links [ edit ] Classification D ICD - 10 : Q87.8 OMIM : 241080 MeSH : C536742 External resources Orphanet : 3464 v t e Disorders involving multiple endocrine glands Autoimmune polyendocrine syndrome APS1 APS2 Carcinoid syndrome Multiple endocrine neoplasia 1 2A 2B Progeria Werner syndrome Acrogeria Metageria Woodhouse–Sakati syndrome
A number sign (#) is used with this entry because of evidence that Woodhouse-Sakati syndrome (WDSKS) is caused by homozygous mutation in the C2ORF37 gene (DCAF17; 612515) on chromosome 2q31. ... Gul et al. (2000) referred to the disorder in the proband as 'Woodhouse and Sakati syndrome' and suggested autosomal recessive inheritance. ... One of the families had been previously reported by Woodhouse and Sakati (1983) and 3 affected members had since developed a neurologic extrapyramidal syndrome with choreoathetoid movements and dystonia. ... Al-Semari and Bohlega (2007) suggested that the disorder could be an undescribed type of neuro-endocrine-ectodermal syndrome. Medica et al. (2007) reported a 52-year-old woman of Croatian origin who had the phenotypic findings of Woodhouse-Sakati syndrome, including hypogonadism, sparse hair, diabetes mellitus, sensorineural hearing impairment, mild mental retardation, and flattened T waves on EKG. Koshy et al. (2008) reported 3 sibs from a consanguineous Indian family with Woodhouse-Sakati syndrome who showed phenotypic variability.
Woodhouse-Sakati syndrome is a multisystemic disorder characterized by hypogonadism, alopecia, diabetes mellitus, intellectual deficit and extrapyramidal signs with choreoathetoid movements and dystonia. ... Additional manifestations may include sensorineural deafness, flattened T waves on ECG, seizures, sensory polyneuropathy, dysarthria, various craniofacial abnormalities (high forehead, flat occiput, triangular face, prominent nasal root, hypertelorism, and down-slanting palpebral fissures), scoliosis, hyperreflexia, and camptodactyly. Etiology Woodhouse-Sakati syndrome is associated with mutations in the DCAF17 gene (2q31.1), encoding a nucleolar protein of unknown function.
Diagnosis Suggestive Findings Woodhouse-Sakati syndrome (WSS) should be suspected in individuals with the following findings. ... The growth pattern is normal and growth hormone levels are usually normal; short stature is not a part of this syndrome [Agopiantz et al 2014]. Diabetes mellitus. ... XL AD AR Idiopathic hypogonadotropic hypogonadism w/congenital olfactory deficit w/ataxia, epilepsy & congenital paresis of cranial nerves III, IV, VI Gordon Holmes syndrome (OMIM 212840) RNF216 PNPLA6 2 AR Hypogonadism, cerebellar ataxia, white matter lesions; no alopecia Alopecia, neuropathy, endocrinopathy (ANE) syndrome (OMIM 612079) 3 RBM28 AR Hypogonadotropic hypogonadism, alopecia, adrenal insufficiency and cognitive impairment Hutchinson-Gilford progeria syndrome LMNA AD 4 Alopecia, aged-looking skin, joint abnormalities, loss of subcutaneous fat Cockayne syndrome ERCC6 ERCC8 AR Short stature & appearance of premature aging Deafness-dystonia-optic neuronopathy syndrome (Mohr-Tranebjaerg syndrome) TIMM8A 5 XL Early-onset hearing loss; movement disorder; impaired vision; behavior problems (almost exclusively in males) Neurodegeneration with brain iron storage disorders PANK2 PLA2G6 C19orf12 FA2H ATP13A2 WDR45 COASY FTL CP AR XL AD 6 Variable phenotype: variable age of onset, dystonia w/postural instability Brain iron accumulation on MRI Dystonia See footnote 7. ... Almost all individuals with Hutchinson-Gilford progeria syndrome have the disorder as the result of a de novo autosomal dominant pathogenic variant. 5. Deafness-dystonia-optic neuronopathy syndrome occurs as either a single-gene disorder resulting from mutation of TIMM8A or a contiguous gene deletion syndrome at Xq22. 6.
Woodhouse-Sakati syndrome is a disorder that primarily affects the body's network of hormone-producing glands (the endocrine system ) and the nervous system. ... As a result, people with Woodhouse-Sakati syndrome usually have an inability to conceive children (infertility). ... For this reason, Woodhouse-Sakati syndrome is sometimes classified as part of a group of disorders called neurodegeneration with brain iron accumulation (NBIA). Frequency Woodhouse-Sakati syndrome is a rare disorder; its prevalence is unknown. Only a few dozen affected families, mostly in the Middle East, have been described in the medical literature. Causes Woodhouse-Sakati syndrome is caused by mutations in the DCAF17 gene.
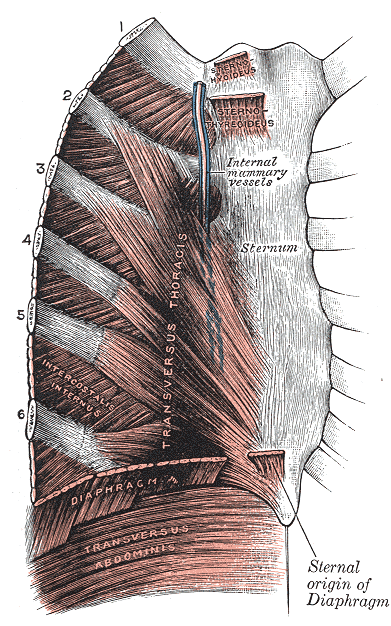
Not to be confused with Tietz syndrome . This article needs additional citations for verification . ... Unsourced material may be challenged and removed. Find sources: "Tietze syndrome" – news · newspapers · books · scholar · JSTOR ( August 2010 ) ( Learn how and when to remove this template message ) Tietze syndrome Other names Chondropathia tuberosa, costochondral junction syndrome Sternocostals and interchondral articulations. Anterior view. ( Costal cartilages visible on diagram.) Specialty Rheumatology Tietze syndrome (also called costochondral junction syndrome ) is a benign inflammation of one or more of the costal cartilages . [1] It was first described in 1921 by the German surgeon Alexander Tietze (1864–1927). [2] [3] Tietze syndrome is not the same as costochondritis . [4] Tietze syndrome is differentiated from costochondritis by swelling of the costal cartilages, which does not appear in costochondritis. ... Berliner klinische Wochenschrift . 58 : 829–831. ^ "Tietze's Syndrome" . ^ Bhat, Sriram (2010). Tietze's disease . ... Retrieved 2018-04-25 . ^ a b "Tietze Syndrome - Symptoms, Causes, Treatment, Pictures" . syndrome.org .
Tietze syndrome is an inflammatory condition characterized by chest pain and swelling of the cartilage around the ribs. Specifically, people with Tietze syndrome have swelling of the cartilage that joins the upper ribs to the breastbone. ... The chest, shoulders, and arms may also have redness and warmth. In some cases, Tietze syndrome may resolve on its own without treatment, while other people experience patterns of pain followed by some relief of pain. ... Your doctor may also recommend seeing a chiropractor . Of note, this syndrome is different from Tietz syndrome , which is characterized by profound hearing loss from birth, fair skin, and light-colored hair.
Costochondritis is sometimes known as chest wall pain syndrome, costosternal syndrome or costosternal chondrodynia. Sometimes, swelling accompanies the pain (Tietze syndrome). What causes costochondritis is unclear. ... Risk factors Costochondritis occurs most often in women older than 40. Tietze syndrome usually occurs in teenagers and young adults, and with equal frequency in men and women.
Human disease Costochondritis The costal cartilages Specialty Emergency medicine Symptoms Pain and, inflammation in the costal cartilage Diagnostic method Based on symptoms after ruling out underlying causes Differential diagnosis heart attack , chest pain , heartburn , Medication NSAIDs Costochondritis, also known as chest wall pain , costosternal syndrome , or costosternal chondrodynia [1] is an acute [2] and often temporary inflammation of the costal cartilage , the structure that connects each rib to the sternum at the costosternal joint . The condition is a common cause of chest pain . [1] [3] [4] Though costochondritis often resolves on its own, it can be a recurring condition that has little or no signs of onset. [5] Costochondritis symptoms can be similar to the chest pain associated with a heart attack . [6] [7] Chest pain is considered a medical emergency until life-threatening heart disease (such as an acute coronary syndrome ) can be ruled out. [4] [7] Severe cases of costal cartilage inflammation that also involve painful swelling are sometimes referred to as Tietze's syndrome , a term sometimes used interchangeably with costochondritis. However, some physicians view costochondritis and Tietze's syndrome as separate disease states due to the absence of costal cartilage swelling in costochondritis. [2] [8] Treatment options for costochondritis are limited and usually involve a combination of rest, pain-relieving medications , and anti-inflammatory medications. [8] Cases with persistent discomfort may be managed with cortisone injections [6] [8] or surgery may be indicated if the condition is severe. ... Retrieved 26 December 2012 . ^ Seferović PM, Ristić AD, Maksimović R, Simeunović DS, Milinković I, Seferović Mitrović JP, Kanjuh V, Pankuweit S, Maisch B (May 2013). "Pericardial syndromes: an update after the ESC guidelines 2004". ... External links [ edit ] Classification D ICD - 10 : M94.0 ICD - 9-CM : 733.6 External resources MedlinePlus : 000164 MayoClinic.com Benign Tender Chest Wall Pain of Costosternal Joints Inflammation at patient.info Costochondritis and Tietze Syndrome