-
Myxoid Liposarcoma
Wikipedia
External links [ edit ] Myxoid liposarcoma: a rare soft-tissue tumor with a misleading benign appearance Efficacy of first-line doxorubicin and ifosfamide in myxoid liposarcoma. 2012 Classification D ICD-O : 8852/3 OMIM : 613488 MeSH : D018208 v t e Connective / soft tissue tumors and sarcomas Not otherwise specified Soft-tissue sarcoma Desmoplastic small-round-cell tumor Connective tissue neoplasm Fibromatous Fibroma / fibrosarcoma : Dermatofibrosarcoma protuberans Desmoplastic fibroma Fibroma / fibromatosis : Aggressive infantile fibromatosis Aponeurotic fibroma Collagenous fibroma Diffuse infantile fibromatosis Familial myxovascular fibromas Fibroma of tendon sheath Fibromatosis colli Infantile digital fibromatosis Juvenile hyaline fibromatosis Plantar fibromatosis Pleomorphic fibroma Oral submucous fibrosis Histiocytoma / histiocytic sarcoma : Benign fibrous histiocytoma Malignant fibrous histiocytoma Atypical fibroxanthoma Solitary fibrous tumor Myxomatous Myxoma / myxosarcoma Cutaneous myxoma Superficial acral fibromyxoma Angiomyxoma Ossifying fibromyxoid tumour Fibroepithelial Brenner tumour Fibroadenoma Phyllodes tumor Synovial -like Synovial sarcoma Clear-cell sarcoma Lipomatous Lipoma / liposarcoma Myelolipoma Myxoid liposarcoma PEComa Angiomyolipoma Chondroid lipoma Intradermal spindle cell lipoma Pleomorphic lipoma Lipoblastomatosis Spindle cell lipoma Hibernoma Myomatous general: Myoma / myosarcoma smooth muscle : Leiomyoma / leiomyosarcoma skeletal muscle : Rhabdomyoma / rhabdomyosarcoma : Embryonal rhabdomyosarcoma Sarcoma botryoides Alveolar rhabdomyosarcoma Leiomyoma Angioleiomyoma Angiolipoleiomyoma Genital leiomyoma Leiomyosarcoma Multiple cutaneous and uterine leiomyomatosis syndrome Multiple cutaneous leiomyoma Neural fibrolipoma Solitary cutaneous leiomyoma STUMP Complex mixed and stromal Adenomyoma Pleomorphic adenoma Mixed Müllerian tumor Mesoblastic nephroma Wilms' tumor Malignant rhabdoid tumour Clear-cell sarcoma of the kidney Hepatoblastoma Pancreatoblastoma Carcinosarcoma Mesothelial Mesothelioma Adenomatoid tumor v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22 This oncology article is a stub .DDIT3, FUS, EWSR1, CD6, PIK3CA, MDM2, LOC110806263, CTAG1B, CTAG1A, MVP, PIK3CB, LRP1, PIK3CG, HCCS, GLI1, TP53, PIK3CD, TERT, CREB3L2, PRG4, ERG, PTPRA, CDK4, ATF1, RPSA, NR4A3, THPO, WNT1, VIM, HMGA2, ZEB1, SYT1, FGF23, EBP, TAF15, CLDN6, CIB1, SSX2, PRAME, SMUG1, INTS1, CTAG2, ZNF654, ANKRD36B, PDIK1L, MIR135B, MXLPO, SS18, AKT1, S100B, S100A1, CD34, CDKN1B, CDKN2A, E2F1, ERCC5, FGFR2, FLT1, MTOR, GDNF, HMGA1, HSP90AA1, HTC2, IGF1, IGF1R, IL4R, MAD2L1, MAGEA3, MXI1, MYC, NNAT, SERPINE1, PGF, ALK, PLAG1, PMAIP1, PML, PPARG, H3P10

-
Central Core Disease
Wikipedia
External links [ edit ] Classification D ICD - 10 : G71.2 ICD - 9-CM : 359.0 OMIM : 117000 MeSH : D020512 External resources eMedicine : neuro/76 GeneReviews : NBK1391 Orphanet : 597 v t e Diseases of muscle , neuromuscular junction , and neuromuscular disease Neuromuscular- junction disease autoimmune Myasthenia gravis Lambert–Eaton myasthenic syndrome Neuromyotonia Myopathy Muscular dystrophy ( DAPC ) AD Limb-girdle muscular dystrophy 1 Oculopharyngeal Facioscapulohumeral Myotonic Distal (most) AR Calpainopathy Limb-girdle muscular dystrophy 2 Congenital Fukuyama Ullrich Walker–Warburg XR dystrophin Becker's Duchenne Emery–Dreifuss Other structural collagen disease Bethlem myopathy PTP disease X-linked MTM adaptor protein disease BIN1-linked centronuclear myopathy cytoskeleton disease Nemaline myopathy Zaspopathy Channelopathy Myotonia Myotonia congenita Thomsen disease Neuromyotonia / Isaacs syndrome Paramyotonia congenita Periodic paralysis Hypokalemic Thyrotoxic Hyperkalemic Other Central core disease Mitochondrial myopathy MELAS MERRF KSS PEO General Inflammatory myopathy Congenital myopathy v t e Diseases of ion channels Calcium channel Voltage-gated CACNA1A Familial hemiplegic migraine 1 Episodic ataxia 2 Spinocerebellar ataxia type-6 CACNA1C Timothy syndrome Brugada syndrome 3 Long QT syndrome 8 CACNA1F Ocular albinism 2 CSNB2A CACNA1S Hypokalemic periodic paralysis 1 Thyrotoxic periodic paralysis 1 CACNB2 Brugada syndrome 4 Ligand gated RYR1 Malignant hyperthermia Central core disease RYR2 CPVT1 ARVD2 Sodium channel Voltage-gated SCN1A Familial hemiplegic migraine 3 GEFS+ 2 Febrile seizure 3A SCN1B Brugada syndrome 6 GEFS+ 1 SCN4A Hypokalemic periodic paralysis 2 Hyperkalemic periodic paralysis Paramyotonia congenita Potassium-aggravated myotonia SCN4B Long QT syndrome 10 SCN5A Brugada syndrome 1 Long QT syndrome 3 SCN9A Erythromelalgia Febrile seizure 3B Paroxysmal extreme pain disorder Congenital insensitivity to pain Constitutively active SCNN1B / SCNN1G Liddle's syndrome SCNN1A / SCNN1B / SCNN1G Pseudohypoaldosteronism 1AR Potassium channel Voltage-gated KCNA1 Episodic ataxia 1 KCNA5 Familial atrial fibrillation 7 KCNC3 Spinocerebellar ataxia type-13 KCNE1 Jervell and Lange-Nielsen syndrome Long QT syndrome 5 KCNE2 Long QT syndrome 6 KCNE3 Brugada syndrome 5 KCNH2 Short QT syndrome KCNQ1 Jervell and Lange-Nielsen syndrome Romano–Ward syndrome Short QT syndrome Long QT syndrome 1 Familial atrial fibrillation 3 KCNQ2 BFNS1 Inward-rectifier KCNJ1 Bartter syndrome 2 KCNJ2 Andersen–Tawil syndrome Long QT syndrome 7 Short QT syndrome KCNJ11 TNDM3 KCNJ18 Thyrotoxic periodic paralysis 2 Chloride channel CFTR Cystic fibrosis Congenital absence of the vas deferens CLCN1 Thomsen disease Myotonia congenita CLCN5 Dent's disease CLCN7 Osteopetrosis A2, B4 BEST1 Vitelliform macular dystrophy CLCNKB Bartter syndrome 3 TRP channel TRPC6 FSGS2 TRPML1 Mucolipidosis type IV Connexin GJA1 Oculodentodigital dysplasia Hallermann–Streiff syndrome Hypoplastic left heart syndrome GJB1 Charcot–Marie–Tooth disease X1 GJB2 Keratitis–ichthyosis–deafness syndrome Ichthyosis hystrix Bart–Pumphrey syndrome Vohwinkel syndrome ) GJB3 / GJB4 Erythrokeratodermia variabilis Progressive symmetric erythrokeratodermia GJB6 Clouston's hidrotic ectodermal dysplasia Porin AQP2 Nephrogenic diabetes insipidus 2 See also: ion channelsRYR1, LMNA, LARGE1, FKTN, FKRP, ITGA7, POMGNT1, POMT1, CHKB, B4GAT1, SLC26A3, RYR2, RUNX2, SELENON, EPHA3, CACNA1S, MYH7, SCN5A, STK11, ADAMTS13, MYOT, BMS1, PRKAA2, SIRT1, TNNI3K, PRKAB1, ACACA, PRKAA1, FKBP1AP1, ALDH2, ARVD3, ATP2A1, DNM2, ELANE, FKBP1A, FKBP1AP2, NFATC1, FKBP1AP3, FKBP1AP4, IL6, IL17A, ACTB, MYL3, MED25

-
Tourette Syndrome
Medlineplus
Tourette syndrome is a complex disorder characterized by repetitive, sudden, and involuntary movements or noises called tics. ... The involuntary use of inappropriate or obscene language (coprolalia) is possible, but uncommon, among people with Tourette syndrome. In addition to frequent tics, people with Tourette syndrome are at risk for associated problems including attention-deficit/hyperactivity disorder (ADHD), obsessive-compulsive disorder (OCD), anxiety, depression, and problems with sleep. Frequency Although the exact incidence of Tourette syndrome is uncertain, it is estimated to affect 1 to 10 in 1,000 children. ... It is unclear how mutations in the SLITRK1 gene can lead to this disorder. Most people with Tourette syndrome do not have a mutation in the SLITRK1 gene. ... Researchers suspect that changes in other genes, which have not been identified, are also associated with Tourette syndrome. Learn more about the gene associated with Tourette syndrome SLITRK1 Inheritance Pattern The inheritance pattern of Tourette syndrome is unclear.SLITRK1, HDC, DRD3, DRD2, CHD2, SLC6A3, MET, COL27A1, LMO3, MGST1, FPR3, NECTIN1, CSMD3, ZNF577, LINC01122, POU1F1, FLT3, IMMP2L, GTS, MCF2L, LRP2, TNF, DRD4, DBH, HTR2A, SLC6A4, TYMS, BDNF, TDO2, ADD2, HTR1A, MAOA, VDR, DRD1, DNTT, COMT, PSD4, CNTNAP2, ARNTL, BTBD9, GH1, IL10, IL1RN, IL1B, HTR3A, TH, ACP1, SLC1A3, SLC6A2, SMN1, SMN2, XRCC1, SGCE, NRXN1, CHPT1, DHDDS, HTR2C, AADAC, CNR1, HLA-A, HTR2B, CNTN6, NLGN4X, SLC4A10, SLC17A7, CCT6A, CD8A, ASH1L, TDP1, TESC, ROBO4, KCNK9, F11R, HPGDS, LHX6, DLGAP3, SLITRK5, CD69, PNKD, CDH2, RPGRIP1L, WWC1, CDX2, COBLL1, OGA, PDLIM5, OLFM1, GRHL3, NTN4, CHAT, TPH2, MIR429, ADHD2, LHX8, MIR24-1, ADORA1, SKOR1, DYT15, HEXD, ANKK1, RLS1, CBLN2, LYPD6, OLIG1, SLC5A7, ADORA2A, PARP1, ADRA1A, NLRP3, ADRA2A, ATAD1, AMELX, APBA2, PARP9, C1QBP, ALG9, ROBO3, OLIG2, HTR3B, PDLIM7, LAMC2, MAP2K5, GDNF, PKM, PEX13, NFE2L2, MOG, GPT, GRIN2B, MEIS1, GSK3B, GSTP1, LCT, KCNJ5, HTR1B, JAK2, ING2, IL12B, IL10RA, HK1, CXCL8, IL2, IL1R1, HLA-DRB1, IL1A, HTR7, HMBS, PVALB, GCH1, SCN2A, SCN3A, CHM, NR4A3, CHRNA4, WAS, TYR, COL8A1, TSC1, TP53, CSF2, DPP6, DRD5, STAT3, ST14, SPINT1, SNCA, TOR1A, EEF1A2, SLC12A3, CELSR3, ENO2, GPC5, GABPA, SLC1A1, SHOX, CCL2, AUTS1
-
Rubinstein-Taybi Syndrome
Medlineplus
Rubinstein-Taybi syndrome is a condition characterized by short stature, moderate to severe intellectual disability, distinctive facial features, and broad thumbs and first toes . ... Abnormal brain development is thought to underlie intellectual disability in people with Rubinstein-Taybi syndrome. Researchers have not determined how CREBBP gene mutations lead to other signs and symptoms of Rubinstein-Taybi syndrome. ... Several cases of severe Rubinstein-Taybi syndrome have resulted from a deletion of genetic material from the short (p) arm of chromosome 16. ... Researchers believe that the loss of multiple genes in this region probably accounts for the serious complications associated with severe Rubinstein-Taybi syndrome. Some researchers suggest that these cases are a separate condition called chromosome 16p13.3 deletion syndrome. ... Nearly 30 to 40 percent of people with Rubinstein-Taybi syndrome do not have an identified mutation in the CREBBP or EP300 gene or a chromosome 16 deletion.
-
Erythema Elevatum Diutinum
Wikipedia
Erythema elevatum diutinum Erythema elevatum diutinum seen on back of hand Specialty Dermatology Erythema elevatum diutinum is a form of vasculitis . [1] : 835 It has been described as a paraneoplastic syndrome. [2] See also [ edit ] Cutaneous small-vessel vasculitis List of cutaneous conditions References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). ... "Erythema elevatum diutinum as a paraneoplastic syndrome in a patient with pulmonary lymphoepithelioma-like carcinoma". ... External links [ edit ] Classification D ICD - 10 : L95.1 ICD - 9-CM : 695.89 MeSH : C535509 C535509, C535509 DiseasesDB : 31378 External resources eMedicine : derm/133 v t e Cutaneous vasculitis and other vascular-related cutaneous conditions Cutaneous vasculitis Erythema elevatum diutinum Capillaritis Urticarial vasculitis Nodular vasculitis Microvascular occlusion Calciphylaxis Cryoglobulinemic purpura / Cryoglobulinemic vasculitis vascular coagulopathy : Livedoid vasculitis Livedoid dermatitis Perinatal gangrene of the buttock Malignant atrophic papulosis Sneddon's syndrome Purpura Nonthrombocytopenic purpura : Cryofibrinogenemic purpura Drug-induced purpura Food-induced purpura IgA vasculitis Obstructive purpura Orthostatic purpura Purpura fulminans Purpura secondary to clotting disorders Purpuric agave dermatitis Pigmentary purpuric eruptions Solar purpura Traumatic purpura Waldenström hyperglobulinemic purpura Painful bruising syndrome ungrouped: Paroxysmal hand hematoma Postcardiotomy syndrome Deep vein thrombosis Superficial thrombophlebitis Mondor's disease Blueberry muffin baby Fibrinolysis syndrome Systemic vasculitis see Template:Systemic vasculitis Vascular malformations Arteriovenous malformation Bonnet–Dechaume–Blanc syndrome Cobb syndrome Parkes Weber syndrome Sinusoidal hemangioma lymphatic malformation Hennekam syndrome Aagenaes syndrome telangiectasia : Generalized essential telangiectasia Hereditary hemorrhagic telangiectasia Unilateral nevoid telangiectasia Ulcer Venous ulcer Arterial insufficiency ulcer Hematopoietic ulcer Neuropathic ulcer Acroangiodermatitis Lymphedema see Template:Lymphatic vessel disease Ungrouped vascular-related cutaneous conditions Raynaud's phenomenon Thromboangiitis obliterans Erythromelalgia Septic thrombophlebitis Arteriosclerosis obliterans Bier spots / Marshall–White syndrome Cholesterol embolus Reactive angioendotheliomatosis Trousseau's syndrome v t e Paraneoplastic syndromes Endocrine Hypercalcaemia SIADH Zollinger–Ellison syndrome Cushing's syndrome Hematological Multicentric reticulohistiocytosis Nonbacterial thrombotic endocarditis Neurological Paraneoplastic cerebellar degeneration Encephalomyelitis Limbic encephalitis Opsoclonus Polymyositis Transverse myelitis Lambert–Eaton myasthenic syndrome Anti-NMDA receptor encephalitis Musculoskeletal Dermatomyositis Hypertrophic osteopathy Mucocutaneous reactive erythema Erythema gyratum repens Necrolytic migratory erythema papulosquamous Acanthosis nigricans Ichthyosis acquisita Acrokeratosis paraneoplastica of Bazex Extramammary Paget's disease Florid cutaneous papillomatosis Leser-Trélat sign Pityriasis rotunda Tripe palms Other Febrile neutrophilic dermatosis Pyoderma gangrenosum Paraneoplastic pemphigus This cutaneous condition article is a stub .
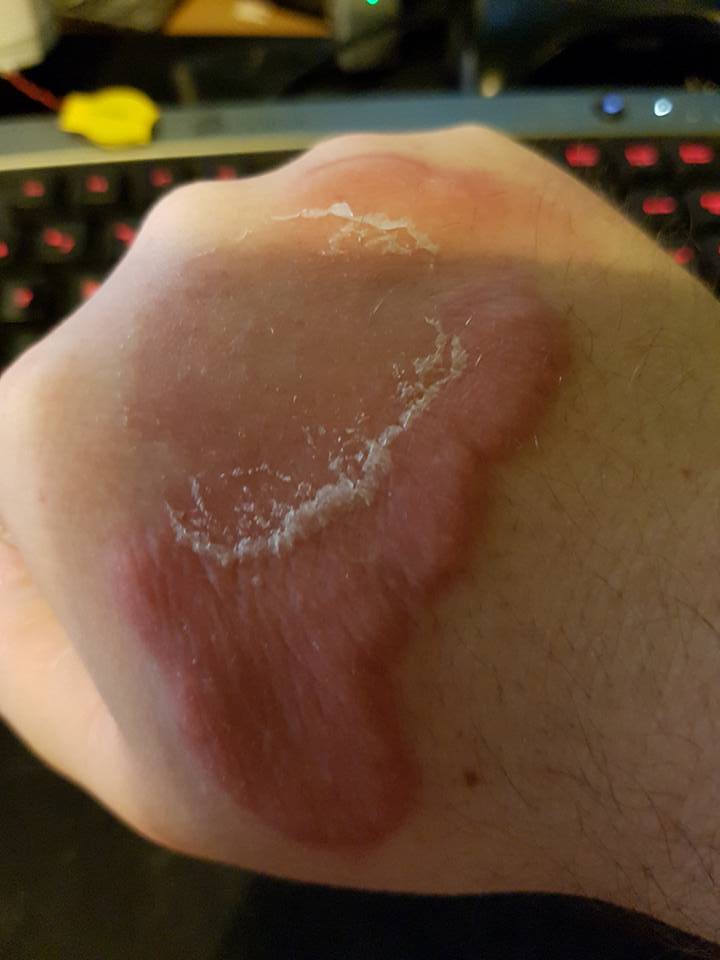
-
Adnp Syndrome
Orphanet
Epidemiology The point prevalence of ADNP syndrome is approximately 1-2/100,000 individuals. The syndrome represents 0.17% of the autism spectrum disorder cases. ... Differential diagnosis Some similarities are found with Okihiro syndrome plus developmental delay, Angelman syndrome, Rett syndrome, Noonan syndrome, Kleefstra syndrome, Smith-Magenis syndrome, and other Coffin-Siris syndrome related disorders. ... Prognosis With developmental delays, it is expected that children with ADNP syndrome will start walking and talking relatively late. ... To date, there is only a paucity of adults that are known to have the ADNP syndrome.

-
Cytochrome B5 Deficiency
Wikipedia
External links [ edit ] Online Mendelian Inheritance in Man (OMIM): 250790 – Methemoglobinemia due to deficiency of cytochrome b 5 v t e Gonadal disorder Ovarian Polycystic ovary syndrome Premature ovarian failure Estrogen insensitivity syndrome Hyperthecosis Testicular Enzymatic 5α-reductase deficiency 17β-hydroxysteroid dehydrogenase deficiency aromatase excess syndrome Androgen receptor Androgen insensitivity syndrome Familial male-limited precocious puberty Partial androgen insensitivity syndrome Other Sertoli cell-only syndrome General Hypogonadism Delayed puberty Hypergonadism Precocious puberty Hypoandrogenism Hypoestrogenism Hyperandrogenism Hyperestrogenism Postorgasmic illness syndrome Cytochrome P450 oxidoreductase deficiency Cytochrome b5 deficiency Androgen-dependent condition Aromatase deficiency Complete androgen insensitivity syndrome Mild androgen insensitivity syndrome Hypergonadotropic hypogonadism Hypogonadotropic hypogonadism Fertile eunuch syndrome Estrogen-dependent condition Premature thelarche Gonadotropin insensitivity Hypergonadotropic hypergonadism v t e Inborn errors of steroid metabolism Mevalonate pathway HMG-CoA lyase deficiency Hyper-IgD syndrome Mevalonate kinase deficiency To cholesterol 7-Dehydrocholesterol path: Hydrops-ectopic calcification-moth-eaten skeletal dysplasia CHILD syndrome Conradi-Hünermann syndrome Lathosterolosis Smith–Lemli–Opitz syndrome desmosterol path: Desmosterolosis Steroids Corticosteroid (including CAH ) aldosterone : Glucocorticoid remediable aldosteronism cortisol / cortisone : CAH 17α-hydroxylase CAH 11β-hydroxylase both: CAH 3β-dehydrogenase CAH 21-hydroxylase Apparent mineralocorticoid excess syndrome/11β-dehydrogenase Sex steroid To androgens 17α-Hydroxylase deficiency 17,20-Lyase deficiency Cytochrome b 5 deficiency 3β-Hydroxysteroid dehydrogenase deficiency 17β-Hydroxysteroid dehydrogenase deficiency 5α-Reductase deficiency Pseudovaginal perineoscrotal hypospadias To estrogens Aromatase deficiency Aromatase excess syndrome Other X-linked ichthyosis Antley–Bixler syndrome This article about a disease , disorder, or medical condition is a stub .
-
Blepharophimosis Intellectual Disability Syndromes
Gard
Blepharophimosis intellectual disability syndromes refers to a group of syndromes, including Ohdo syndrome and Say Barber Biesecker Young-Simpson syndrome, that are characterized by narrow eye openings (blepharophimosis), drooping of the upper eye lids (ptosis) and intellectual disability. ... Ohdo published an article in 1986 describing the first three cases of this syndrome (in a brother, sister, and cousin). Since that time many other case reports describing people with similar (yet variable) features have been reported. The syndromes usually occur sporadically . In most cases the underlying cause can not be determined.
-
Gitelman Syndrome
Omim
For a discussion of genetic heterogeneity of Bartter syndrome, see 607364. Clinical Features It has been proposed that Bartter syndrome, defined generically as an autosomal recessive disorder featuring hypokalemic metabolic alkalosis with salt wasting, is a heterogeneous entity with at least 2 subsets, Gitelman syndrome and 'true Bartter syndrome.' ... Because all of the reports of an association with Bartter syndrome referred to hypomagnesemia and hypocalciuria, Hisakawa et al. (1998) suggested that these might be cases of Gitelman syndrome, not 'true' Bartter syndrome. ... Gitelman syndrome is widely described as a benign or milder variant of Bartter syndrome. ... Clinical presentation included episodes of dehydration, weakness, and failure to thrive, much more suggestive of classic Bartter syndrome type 3 (607364) than of Gitelman syndrome. ... In the follow-up, a drop of both parameters below normal range was a consistent finding reflecting a transition from classic Bartter syndrome to Gitelman syndrome phenotype.SLC12A3, CLCNKB, WNK4, STK39, REN, KCNJ1, TESC, TSC1, PTH, AGT, ACTB, IGAN1, TRPV5, CABIN1, BEST1, RGS2, SLC12A1, SCN5A, PVALB, PTGS2, NOS3, PPP1R12A, DMD, DBP, CASR, ARHGEF25
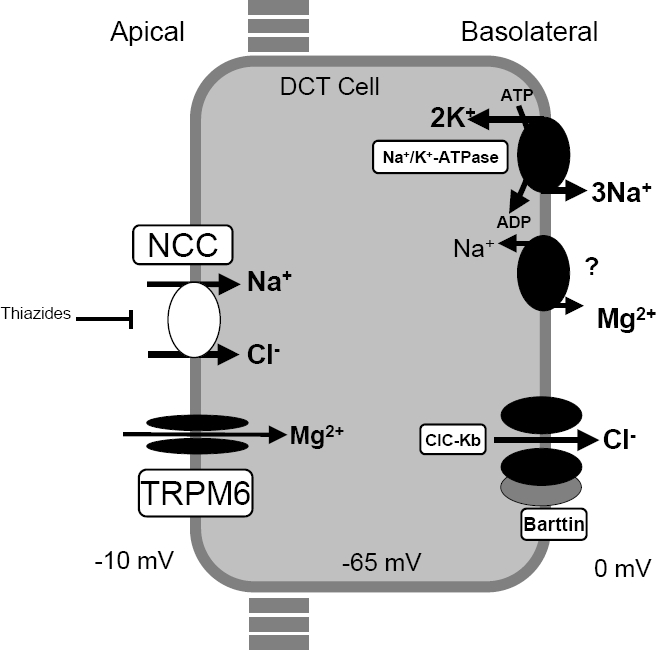
-
Pronator Teres Syndrome
Wikipedia
Pronator teres syndrome Median nerve displayed Specialty Neurology Pronator teres syndrome is a compression neuropathy of the median nerve at the elbow. It is rare compared to compression at the wrist ( carpal tunnel syndrome ) or isolated injury of the anterior interosseous branch of the median nerve ( anterior interosseous syndrome ). ... "Median nerve entrapment syndrome in the proximal forearm." The Journal of Hand Surgery 4, no. 1 (January 1979): 48-51. ... "The pronator teres syndrome: compressive neuropathy of the median nerve." ... "MR imaging features of radial tunnel syndrome: initial experience." Radiology 240, no. 1 (July 2006): 161-8.

-
Genetic Disorder
Wikipedia
Some X-linked dominant conditions, such as Rett syndrome , incontinentia pigmenti type 2, and Aicardi syndrome , are usually fatal in males either in utero or shortly after birth, and are therefore predominantly seen in females. ... An example of these disorders is trisomy 21 ( Down syndrome ), in which there is an extra copy of chromosome 21. ... Many genetic disorders affect stages of development, such as Down syndrome , while others result in purely physical symptoms such as muscular dystrophy . ... Retrieved 2019-07-01 . ^ Keane MG; Pyeritz RE (May 2008). "Medical management of Marfan syndrome" . Circulation . 117 (21): 2802–13. doi : 10.1161/CIRCULATIONAHA.107.693523 . ... External links [ edit ] Classification D MeSH : D030342 DiseasesDB : 28838 Public Health Genomics at CDC OMIM — Online Mendelian Inheritance in Man, a catalog of human genes and genetic disorders Genetic and Rare Diseases Information Center (GARD) Office of Rare Diseases (ORD), National Institutes of Health (NIH) CDC’s National Center on Birth Defects and Developmental Disabilities Genetic Disease Information from the Human Genome Project Global Genes Project, Genetic and Rare Diseases Organization List of Genetic Disorders - Genome.gov v t e Personal genomics Data collection Biobank Biological database Field concepts Biological specimen De-identification Human genetic variation Genetic linkage Single-nucleotide polymorphisms Identity by descent Genetic disorder Applications Personalized medicine Predictive medicine Genetic epidemiology Pharmacogenomics Analysis techniques Whole genome sequencing Genome-wide association study SNP array Genetic testing Major projects Human Genome Project International HapMap Project 1000 Genomes Project Human Genome Diversity Project v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions ) v t e Genetic disorder , membrane: Solute carrier disorders 1-10 SLC1A3 Episodic ataxia 6 SLC2A1 De Vivo disease SLC2A5 Fructose malabsorption SLC2A10 Arterial tortuosity syndrome SLC3A1 Cystinuria SLC4A1 Hereditary spherocytosis 4 / Hereditary elliptocytosis 4 SLC4A11 Congenital endothelial dystrophy type 2 Fuchs' dystrophy 4 SLC5A1 Glucose-galactose malabsorption SLC5A2 Renal glycosuria SLC5A5 Thyroid dyshormonogenesis type 1 SLC6A19 Hartnup disease SLC7A7 Lysinuric protein intolerance SLC7A9 Cystinuria 11-20 SLC11A1 Crohn's disease SLC12A3 Gitelman syndrome SLC16A1 HHF7 SLC16A2 Allan–Herndon–Dudley syndrome SLC17A5 Salla disease SLC17A8 DFNA25 21-40 SLC26A2 Multiple epiphyseal dysplasia 4 Achondrogenesis type 1B Recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia SLC26A4 Pendred syndrome SLC35C1 CDOG 2C SLC39A4 Acrodermatitis enteropathica SLC40A1 African iron overload see also solute carrier family v t e Cell surface receptor deficiencies G protein-coupled receptor (including hormone ) Class A TSHR ( Congenital hypothyroidism 1 ) LHCGR ( Luteinizing hormone insensitivity , Leydig cell hypoplasia , Male-limited precocious puberty ) FSHR ( Follicle-stimulating hormone insensitivity , XX gonadal dysgenesis ) GnRHR ( Gonadotropin-releasing hormone insensitivity ) EDNRB ( ABCD syndrome , Waardenburg syndrome 4a , Hirschsprung's disease 2 ) AVPR2 ( Nephrogenic diabetes insipidus 1 ) PTGER2 ( Aspirin-induced asthma ) Class B PTH1R ( Jansen's metaphyseal chondrodysplasia ) Class C CASR ( Familial hypocalciuric hypercalcemia ) Class F FZD4 ( Familial exudative vitreoretinopathy 1 ) Enzyme-linked receptor (including growth factor ) RTK ROR2 ( Robinow syndrome ) FGFR1 ( Pfeiffer syndrome , KAL2 Kallmann syndrome ) FGFR2 ( Apert syndrome , Antley–Bixler syndrome , Pfeiffer syndrome , Crouzon syndrome , Jackson–Weiss syndrome ) FGFR3 ( Achondroplasia , Hypochondroplasia , Thanatophoric dysplasia , Muenke syndrome ) INSR ( Donohue syndrome Rabson–Mendenhall syndrome ) NTRK1 ( Congenital insensitivity to pain with anhidrosis ) KIT ( KIT Piebaldism , Gastrointestinal stromal tumor ) STPK AMHR2 ( Persistent Müllerian duct syndrome II ) TGF beta receptors : Endoglin / Alk-1 / SMAD4 ( Hereditary hemorrhagic telangiectasia ) TGFBR1 / TGFBR2 ( Loeys–Dietz syndrome ) GC GUCY2D ( Leber's congenital amaurosis 1 ) JAK-STAT Type I cytokine receptor : GH ( Laron syndrome ) CSF2RA ( Surfactant metabolism dysfunction 4 ) MPL ( Congenital amegakaryocytic thrombocytopenia ) TNF receptor TNFRSF1A ( TNF receptor associated periodic syndrome ) TNFRSF13B ( Selective immunoglobulin A deficiency 2 ) TNFRSF5 ( Hyper-IgM syndrome type 3 ) TNFRSF13C ( CVID4 ) TNFRSF13B ( CVID2 ) TNFRSF6 ( Autoimmune lymphoproliferative syndrome 1A ) Lipid receptor LRP : LRP2 ( Donnai–Barrow syndrome ) LRP4 ( Cenani–Lenz syndactylism ) LRP5 ( Worth syndrome , Familial exudative vitreoretinopathy 4 , Osteopetrosis 1 ) LDLR ( LDLR Familial hypercholesterolemia ) Other/ungrouped Immunoglobulin superfamily : AGM3, 6 Integrin : LAD1 Glanzmann's thrombasthenia Junctional epidermolysis bullosa with pyloric atresia EDAR ( EDAR hypohidrotic ectodermal dysplasia ) PTCH1 ( Nevoid basal-cell carcinoma syndrome ) BMPR1A ( BMPR1A juvenile polyposis syndrome ) IL2RG ( X-linked severe combined immunodeficiency ) See also cell surface receptors v t e Diseases of ion channels Calcium channel Voltage-gated CACNA1A Familial hemiplegic migraine 1 Episodic ataxia 2 Spinocerebellar ataxia type-6 CACNA1C Timothy syndrome Brugada syndrome 3 Long QT syndrome 8 CACNA1F Ocular albinism 2 CSNB2A CACNA1S Hypokalemic periodic paralysis 1 Thyrotoxic periodic paralysis 1 CACNB2 Brugada syndrome 4 Ligand gated RYR1 Malignant hyperthermia Central core disease RYR2 CPVT1 ARVD2 Sodium channel Voltage-gated SCN1A Familial hemiplegic migraine 3 GEFS+ 2 Febrile seizure 3A SCN1B Brugada syndrome 6 GEFS+ 1 SCN4A Hypokalemic periodic paralysis 2 Hyperkalemic periodic paralysis Paramyotonia congenita Potassium-aggravated myotonia SCN4B Long QT syndrome 10 SCN5A Brugada syndrome 1 Long QT syndrome 3 SCN9A Erythromelalgia Febrile seizure 3B Paroxysmal extreme pain disorder Congenital insensitivity to pain Constitutively active SCNN1B / SCNN1G Liddle's syndrome SCNN1A / SCNN1B / SCNN1G Pseudohypoaldosteronism 1AR Potassium channel Voltage-gated KCNA1 Episodic ataxia 1 KCNA5 Familial atrial fibrillation 7 KCNC3 Spinocerebellar ataxia type-13 KCNE1 Jervell and Lange-Nielsen syndrome Long QT syndrome 5 KCNE2 Long QT syndrome 6 KCNE3 Brugada syndrome 5 KCNH2 Short QT syndrome KCNQ1 Jervell and Lange-Nielsen syndrome Romano–Ward syndrome Short QT syndrome Long QT syndrome 1 Familial atrial fibrillation 3 KCNQ2 BFNS1 Inward-rectifier KCNJ1 Bartter syndrome 2 KCNJ2 Andersen–Tawil syndrome Long QT syndrome 7 Short QT syndrome KCNJ11 TNDM3 KCNJ18 Thyrotoxic periodic paralysis 2 Chloride channel CFTR Cystic fibrosis Congenital absence of the vas deferens CLCN1 Thomsen disease Myotonia congenita CLCN5 Dent's disease CLCN7 Osteopetrosis A2, B4 BEST1 Vitelliform macular dystrophy CLCNKB Bartter syndrome 3 TRP channel TRPC6 FSGS2 TRPML1 Mucolipidosis type IV Connexin GJA1 Oculodentodigital dysplasia Hallermann–Streiff syndrome Hypoplastic left heart syndrome GJB1 Charcot–Marie–Tooth disease X1 GJB2 Keratitis–ichthyosis–deafness syndrome Ichthyosis hystrix Bart–Pumphrey syndrome Vohwinkel syndrome ) GJB3 / GJB4 Erythrokeratodermia variabilis Progressive symmetric erythrokeratodermia GJB6 Clouston's hidrotic ectodermal dysplasia Porin AQP2 Nephrogenic diabetes insipidus 2 See also: ion channels v t e Genetic disorder , organelle: Peroxisomal disorders and lysosomal structural disorders Peroxisome biogenesis disorder Zellweger syndrome Neonatal adrenoleukodystrophy Infantile Refsum disease Adult Refsum disease-2 RCP 1 Enzyme-related Acatalasia RCP 2&3 Mevalonate kinase deficiency D-bifunctional protein deficiency Adult Refsum disease-1 Transporter-related X-linked adrenoleukodystrophy Lysosomal Danon disease See also: proteins , intermediates v t e Diseases of cilia Structural receptor: Polycystic kidney disease cargo: Asphyxiating thoracic dysplasia basal body : Bardet–Biedl syndrome mitotic spindle : Meckel syndrome centrosome : Joubert syndrome Signaling Nephronophthisis Other/ungrouped Alström syndrome Primary ciliary dyskinesia Senior–Løken syndrome Orofaciodigital syndrome 1 McKusick–Kaufman syndrome Autosomal recessive polycystic kidney See also: ciliary proteins v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteins

-
Usher Syndrome, Type Iiia
Omim
For a discussion of phenotypic heterogeneity of Usher syndrome, see USH1 (276900). Genetic Heterogeneity of Usher syndrome Type III Usher syndrome type IIIB (614504) is caused by mutation in the HARS gene (142810) on chromosome 5q31.3. ... Aller et al. (2004) did not consider progressive hearing loss to be the definitive parameter in distinguishing Usher syndrome type III from Usher syndrome types I and II. ... Mapping In Finnish families segregating Usher syndrome type III, Sankila et al. (1994, 1995) excluded previous chromosomal sites at which Usher syndrome had been mapped. ... In a cohort of 40 Ashkenazi Jewish patients with Usher syndrome, Ness et al. (2003) found that the 16 (40%) who were clinically classified as having Usher syndrome type III were homozygous for the N48K mutation. ... They noted that only 1 other mutation had been reported in the USH3A gene in Spanish families with Usher syndrome (606397.0006). These 2 families accounted for only 1.7% of Spanish families with Usher syndrome.MYO7A, PCDH15, HARS1, PLD4, USH1C, ADGRV1, USH2A, CDH23, CEP250, ARSG, PROM1, CLRN1, ZDHHC24, CDH23-AS1, TRNS2, BBS1, GUCA1A, WHRN, USH1G, PDZD7, CIB2, GJB2, ESPN, HTC2, GC, CEP78, ABHD12, PCARE, GPR166P, LGR6, INTS2, VN1R17P, MRGPRX1, FADS6, MRGPRX3, MRGPRX4, ASIC5, GPR151, OXER1, LCA5, GPRC6A, VEZT, ACTB, MYO15A, PRPH2, ENG, FGF3, GBX2, MEF2C, MYO5B, NOTCH2, NUCB2, OMP, PEX6, RCVRN, RPGR, PHPT1, SOX3, STATH, USH1E, WFS1, FZD4, OTOF, CIB1, ASAH1, NINL, LPAR3, IMMT
-
Three M Syndrome 1
Omim
Other skeletal manifestations include joint hypermobility, joint dislocation, winged scapulae, and pes planus (summary by Badina et al., 2011). Genetic Heterogeneity of 3M Syndrome Also see 3M syndrome-2 (3M2; 612921), caused by mutation in the OBSL1 gene (610991) on chromosome 2q35, and 3M syndrome-3 (3M3; 614205), caused by mutation in the CCDC8 gene (614145) on chromosome 19q13. ... Garcia-Cruz and Cantu (1979) noted mild features of 3M syndrome in presumed heterozygotes. Feldmann et al. (1989) described 2 additional sibs with the 3M syndrome and discussed the differential diagnosis, with particular emphasis on the Russell-Silver syndrome (268650). ... Badina et al. (2011) recommended that patients with 3M syndrome be referred to a pediatric orthopedic surgeon soon after birth. Yakut Short Stature Syndrome Maksimova et al. (2007) identified 43 patients from 37 Yakut families with a short stature syndrome similar to 3M syndrome. ... History Le Merrer et al. (1991) discussed overlap of the 'gloomy face syndrome' with the 3M syndrome. Identity of the 3M syndrome and the 'gloomy face syndrome' was supported by clinical and radiographic long-term follow-up and was established by the demonstration by Huber et al. (2005) of mutations in the CUL7 gene in patients with either diagnosis.
-
Uniparental Disomy
Wikipedia
The most well-known conditions include Prader–Willi syndrome and Angelman syndrome . Both of these disorders can be caused by UPD or other errors in imprinting involving genes on the long arm of chromosome 15 . [6] Other conditions, such as Beckwith–Wiedemann syndrome , are associated with abnormalities of imprinted genes on the short arm of chromosome 11 . ... Retrieved 29 February 2016 . ^ Angelman Syndrome, Online Mendelian Inheritance in Man ^ "OMIM Entry - # 608149 - KAGAMI-OGATA SYNDROME" . omim.org . ... "Chromosome 14 uniparental disomy syndrome information Diseases Database" . www.diseasesdatabase.com . ... National Library of Medicine v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22

-
Juberg-Hayward Syndrome
Wikipedia
Juberg-Hayward syndrome Other names Cleft lip/palate with abnormal thumbs and microcephaly [1] Juberg-Hayward syndrome is a rare genetic syndrome characterised by cleft lip and cleft palate , microcephaly , ptosis, short stature , hypoplasia or aplasia of thumbs, dislocation of radial head and fusion of humerus and radius . ... Contents 1 Presentation 2 Genetics 3 History 4 References 5 External links Presentation [ edit ] These include [2] Growth retardation Microcephaly Cleft lip and palate Minor vertebral and rib anomalies Horseshoe kidneys Thumb anomalies Triphalangeal thumb Radial ray anomalies Genetics [ edit ] This syndrome is caused by mutations in the establishment of cohesion 1 homolog 2 ( ESCO2 ) gene. [3] This gene is located on the short arm of chromosome 8 (8p21.1). Mutations in this gene also cause Roberts/SC phocomelia syndrome . [ citation needed ] Juberg-Hayward syndrome is inherited in both an autosomal recessive and autosomal dominant fashion. [ citation needed ] History [ edit ] This condition was first described in 1969 by Juberg and Hayward. [4] References [ edit ] ^ "Juberg-Hayward syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . Retrieved 20 July 2020 . ^ Verloes A, Le Merrer M , Davin J-C Briard ML, et al (1992) The orocraniodigital syndrome of Juberg and Hayward. J Med Gen 29(4):262-265 ^ Kantaputra PN, Dejkhamron P, Intachai W, Ngamphiw C, Kawasaki K, Ohazama A, Krisanaprakornkit S, Olsen B, Tongsima S, Ketudat Cairns JR (2020) Juberg-Hayward syndrome is a cohesinopathy, caused by mutation in ESCO2. Eur J Orthod ^ Juberg RC, Hayward JR (1969) A new familial syndrome of oral, cranial, and digital anomalies.
-
Environmental Dependency Syndrome
Wikipedia
Zelig syndrome Environmental dependency syndrome , [1] also called Zelig syndrome or Zelig-like syndrome from the name of the protagonist of Woody Allen 's Zelig , [2] is a syndrome where the affected individual relies on environmental cues in order to accomplish goals or tasks. ... Quotes . ^ Karlinsky, Harry (October 2007) [ 1983 ]. " Zelig : Woody Allen's classic film continues to impact the world of psychiatry [Zelig syndrome or Zelig-like syndrome]" . Canadian Psychiatric Association . 3 (5). ... Part II: Patient behavior in complex and social situations: The "environmental dependency syndrome " ". Annals of Neurology . John Wiley & Sons . 19 (4): 335–343. doi : 10.1002/ana.410190405 . ... "On a peculiar environmental dependency syndrome in a case with frontal-temporal damage: Zelig-like syndrome".
-
Lucey–driscoll Syndrome
Wikipedia
Please help to improve this article by introducing more precise citations. ( September 2011 ) ( Learn how and when to remove this template message ) ( Learn how and when to remove this template message ) Lucey–Driscoll syndrome Other names Transient familial neonatal hyperbilirubinemia Lucey–Driscoll syndrome has an autosomal recessive pattern of inheritance. Specialty DiseasesDB = 32677 Lucey–Driscoll syndrome is an autosomal recessive metabolic disorder affecting enzymes involved in bilirubin metabolism. [1] It is one of several disorders classified as a transient familial neonatal unconjugated hyperbilirubinemia . ... Genetics [ edit ] A defect in the UGT1A1 -gene, also linked to Crigler–Najjar syndrome and Gilbert's syndrome , is responsible for the congenital form of Lucey–Driscoll syndrome. ... You can help by adding to it . ( August 2017 ) References [ edit ] ^ "Lucey-Driscoll syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ... External links [ edit ] Classification D ICD - 10 : P59.8 ICD - 9-CM : 774.30 OMIM : 237900 MeSH : C562692 External resources Orphanet : 2312 Online Mendelian Inheritance in Man (OMIM): 237900 - transient familial neonatal hyperbilirubinemia, breast feeding jaundice included v t e Heme metabolism disorders Porphyria , hepatic and erythropoietic ( porphyrin ) early mitochondrial: ALAD porphyria Acute intermittent porphyria cytoplasmic: Gunther disease/congenital erythropoietic porphyria Porphyria cutanea tarda / Hepatoerythropoietic porphyria late mitochondrial: Hereditary coproporphyria Harderoporphyria Variegate porphyria Erythropoietic protoporphyria Hereditary hyperbilirubinemia ( bilirubin ) unconjugated: Gilbert's syndrome Crigler–Najjar syndrome Lucey–Driscoll syndrome conjugated: Dubin–Johnson syndrome nd sheet Rotor syndrome This genetic disorder article is a stub .

-
Burning Feet Syndrome
Wikipedia
Unsourced material may be challenged and removed. Find sources: "Burning feet syndrome" – news · newspapers · books · scholar · JSTOR ( August 2018 ) ( Learn how and when to remove this template message ) Burning feet syndrome Specialty Endocrinology Burning feet syndrome , also known as Grierson-Gopalan syndrome , is a medical condition that causes severe burning and aching of the feet, hyperesthesia , and vasomotor changes of the feet that lead to excessive sweating . ... Causes [ edit ] Burning feet syndrome can be inherited, or it can be caused by pressure being put on the nerves . Links also exist between this syndrome and diseases such as hypothyroidism , diabetes mellitus , and rheumatoid arthritis ; links are also believed to exist between this syndrome and vitamin B (specifically pantothenic acid ) deficiencies and kidney failure . ... Eponym [ edit ] Termed Grierson-Gopalan syndrome after Coluther Gopalan and J. Grierson. [2] [3] [4] See also [ edit ] Erythromelalgia Small fiber peripheral neuropathy Tarsal tunnel syndrome Chemotherapy-induced acral erythema References [ edit ] ^ Tavee J, Zhou L. ... PMID 19414545 [PubMed - indexed for MEDLINE] ^ Grierson-Gopalan syndrome at Who Named It? ^ J. Grierson.
-
Dressler Syndrome
Wikipedia
Not to be confused with DRESS Syndrome . Dressler syndrome Specialty Cardiology Dressler syndrome is a secondary form of pericarditis that occurs in the setting of injury to the heart or the pericardium (the outer lining of the heart). It consists of fever, pleuritic pain, pericarditis and/or a pericardial effusion. Dressler syndrome is also known as postmyocardial infarction syndrome [1] and the term is sometimes used to refer to post-pericardiotomy pericarditis . ... Treatment [ edit ] Dressler syndrome is best treated with high dose aspirin. ... "The post-myocardial-infarction syndrome: a report on forty-four cases". ... "Cardiac tamponade in Dressler's syndrome. Case report". Cardiology . 70 (1): 31–6. doi : 10.1159/000173566 .
-
Occipital Horn Syndrome
Wikipedia
Occipital horn syndrome Other names Ehlers-Danlos syndrome type IX; X-Linked Cutis Laxa This condition is inherited in an X-linked recessive manner. Specialty Endocrinology Complications Aortic aneurysms Medication Droxidopa , copper-histidine injections Occipital horn syndrome ( OHS ), formerly considered a variant of Ehlers–Danlos syndrome , [1] is an X-linked recessive mitochondrial and connective tissue disorder . ... "Chapter 14: Menkes Disease and the Occipital Horn Syndrome". In Royce PM, Steinmann B (eds.). ... October 2017. ^ a b Yasmeen S, Lund K, De Paepe A, De Bie S, Heiberg A, Silva J, Martins M, Skjørringe T, Møller LB (April 2014). "Occipital horn syndrome and classical Menkes Syndrome caused by deep intronic mutations, leading to the activation of ATP7A pseudo-exon" . ... External links [ edit ] Classification D ICD - 10 : E83.0 OMIM : 304150 MeSH : C537860 C537860, C537860 DiseasesDB : 33413 External resources Orphanet : 198 GeneReviews/NCBI/NIH/UW entry on ATP7A-Related Copper Transport Disorders Occipital horn syndrome at NIH 's Office of Rare Diseases v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia v t e Metal deficiency and toxicity disorders Iron excess: Iron overload Hemochromatosis Hemochromatosis/HFE1 Juvenile/HFE2 HFE3 African iron overload/HFE4 Aceruloplasminemia Atransferrinemia Hemosiderosis deficiency: Iron deficiency Copper excess: Copper toxicity Wilson's disease deficiency: Copper deficiency Menkes disease / Occipital horn syndrome Zinc excess: Zinc toxicity deficiency: Acrodermatitis enteropathica Other Inborn errors of metabolismATP7A, PGK1, CENPJ, LOX, COPD, PDAP1, SLC35G1, MTPAP, ASAP1, DESI1, PART1, TUSC2, ACP3, PAPOLA, ATOX1, ASAP2, POMC, REG3A, NOS3, HSPA5, FBN2, MRPS30










