Congenital hepatic fibrosis Congenital hepatic fibrosis has an autosomal recessive pattern of inheritance. Specialty Gasteroenterology Congenital hepatic fibrosis is an inherited fibrocystic liver disease associated with proliferation of interlobular bile ducts within the portal areas and fibrosis that do not alter hepatic lobular architecture. The fibrosis would affect resistance in portal veins leading to portal hypertension . Contents 1 Cause 2 Mechanism 3 Diagnosis 4 Management 5 See also 6 References 7 External links Cause [ edit ] The condition is usually congenital, but sporadic cases have also been reported. It may be associated with other congenital defects, commonly with autosomal recessive polycystic kidney disease , the most severe form of PKD.
Membranoproliferative glomerulonephritis (MPGN) is a chronic progressive kidney disorder characterized by glomerular capillary wall structural changes and mesangial cell proliferation leading to nephrotic syndrome, hypocomplementemia, hypertension, proteinuria and end-stage kidney disease.
A number sign (#) is used with this entry because CFHR5 deficiency is caused by heterozygous mutation in the CFHR5 gene (608593) on chromosome 1q32. Clinical Features Gale et al. (2010) reported 2 unrelated families with an autosomal dominant form of glomerulonephritis resulting in renal failure. Both families had ancestors from the Troodos mountains of Cyprus. Additional patients of Cypriot origin with a similar disorder were subsequently identified. In all, there were 26 patients from 11 families. All had microscopic hematuria, and many developed macroscopic hematuria following upper respiratory tract infections. Renal biopsies showed glomerulonephritis with C3 (120700) deposits in the subendothelium and mesangium.
Description Habib et al. (1973) recognized 2 morphologic classes for the glomerular changes seen in patients with mesangiocapillary (membranoproliferative) glomerulonephritis (MPGN). Type I is characterized by double contour appearance of the capillary walls due to mesangial cell interposition, with nonargyrophilic subendothelial deposits which are finely granular on electron microscopy. Type II is characterized by linear dense deposits within the basement membrane and only rare double contours. These 2 types appear to be distinct with no conversion of one type to another on serial biopsy. Strife et al. (1977) described a third variety in which there are not only subendothelial deposits but also numerous subepithelial and intramembranous deposits, associated with replication of the lamina densa and frequently disruption of the whole basement membrane.
MPGN accounts for approximately 4% of primary renal causes of nephrotic syndrome in children and 7% in adults. [3] It should not be confused with membranous glomerulonephritis , a condition in which the basement membrane is thickened, but the mesangium is not. ... It also is related to a number of autoimmune diseases, prominently systemic lupus erythematosus (SLE). Also found with Sjögren syndrome , rheumatoid arthritis , inherited complement deficiencies (esp C2 deficiency), scleroderma , Celiac disease . [16] The histomorphologic differential diagnosis includes transplant glomerulopathy and thrombotic microangiopathies . ... Membranoproliferative_GN at Nephropathology tutorial MP GN Pathophysiology discusses the nephritic auto-antibodies/factors v t e Disease of the kidney glomerules Primarily nephrotic Non-proliferative Minimal change Focal segmental Membranous Proliferative Mesangial proliferative Endocapillary proliferative Membranoproliferative/mesangiocapillary By condition Diabetic Amyloidosis Primarily nephritic , RPG Type I RPG / Type II hypersensitivity Goodpasture syndrome Type II RPG / Type III hypersensitivity Post-streptococcal Lupus diffuse proliferative IgA Type III RPG / Pauci-immune Granulomatosis with polyangiitis Microscopic polyangiitis Eosinophilic granulomatosis with polyangiitis General glomerulonephritis glomerulonephrosis
In addition to common neurological signs due to mass effect (headache and/or visual field deterioration), additional clinical manifestations include menstrual irregularities (secondary amenorrhea, oligomenorhea or severe menorrhagia), galactorrhea, infertility or ovarian hyperstimulation syndrome (in premenopausal women), testicular enlargement and, occasionally, hypogonadism (in men) and isosexual precocious puberty (in children).
X-linked cleft palate and ankyloglossia is a rare, genetic developmental defect during embryogenesis syndrome characterized by the association of complete, partial or submucous cleft palate and ankyloglossia.
Both the canine and the murine disorders suggest one of the otopalatodigital syndromes (311300, 303400). Pauws et al. (2009) generated a Tbx22-null mouse, which demonstrated a submucous cleft palate (SMCP) and ankyloglossia, similar to the human phenotype, with a small minority showing overt clefts.
Symptoms improve after a few minutes of rest and may be exacerbated by cold. The term Brody syndrome refers to a clinically distinguishable subset of patients without ATP2A1 mutations, with adolescence or adult onset and selective muscular involvement, in which myalgia is more common.
A few episodes of carpopedal spasms had been observed, leading to the diagnosis of Gitelman syndrome (263800). His brother's earliest recollection of a disorder of muscle function was his inability to run in races at the age of 4 to 5 years.
Brody myopathy is a condition that affects the skeletal muscles, which are the muscles used for movement. Affected individuals experience muscle cramping and stiffening after exercise or other strenuous activity, especially in cold temperatures. These symptoms typically begin in childhood. They are usually painless, but in some cases can cause mild discomfort. The muscles usually relax after a few minutes of rest. Most commonly affected are the muscles of the arms, legs, and face (particularly the eyelids). In some people with Brody myopathy, exercise leads to the breakdown of muscle tissue (rhabdomyolysis).
They use the disease term "Brody disease" for individuals with an identified mutation versus "Brody syndrome" for those that do not. More research may help clarify whether these are two different disorders or a variation of the same disorder.
The kidneys are most commonly affected (clinically manifesting as nephrotic syndrome and renal failure), but liver, heart, peripheral nerves, blood vessels, and joints may also be involved.
Progressive myoclonic epilepsy with dystonia is a rare, genetic epilepsy syndrome characterized by neonatal or early infantile onset of severe, progressive, typically frequent and prolonged myoclonic seizures that are refractory to treatment, associated with localized and/or generalized paroxysmal dystonia (which later becomes persistent).
A number sign (#) is used with this entry because early infantile epileptic encephalopathy-16 (EIEE16) is caused by homozygous or compound heterozygous mutation in the TBC1D24 gene (613577) on chromosome 16p13. Mutation in the TBC1D24 gene can also cause familial infantile myoclonic epilepsy (FIME; 605021), a less severe disorder. Description Early infantile epileptic encephalopathy-16 is a severe autosomal recessive neurologic disorder characterized by onset of seizures in the first weeks or months of life. Seizures can be of various types, are unresponsive to medication, last for long periods of time, and occur frequently. Affected infants show psychomotor regression or lack of psychomotor development, as well as other neurologic features such as extrapyramidal signs and hypotonia.
A rare, genetic, lypmhoproliferative syndrome characterized by early onset recurrent infections, lymphadenopathy with hepatosplenomegaly and variabe autoimmune disorders, including hemolytic anemia, thrombocytopenia, neutropenia, enteropathy, type I diabetes, scleroderma, arthritis, atopic dermatitis, and inflammatory lung disease.
A number sign (#) is used with this entry because infantile-onset multisystem autoimmune disease-1 (ADMIO1) is caused by heterozygous mutation in the STAT3 gene (102582) on chromosome 17q21. Description Infantile-onset multisystem autoimmune disease-1 is characterized by early childhood onset of a spectrum of autoimmune disorders affecting multiple organs. Common manifestations include insulin-dependent diabetes mellitus and autoimmune enteropathy, or celiac disease, and autoimmune hematologic disorders. Other features include short stature and nonspecific dermatitis. More variable features include hypothyroidism, autoimmune arthritis, and delayed puberty. Some patients may show recurrent infections. The disorder results from an inborn error of cytokine signaling (summary by Flanagan et al., 2014 and Milner et al., 2015).
Animal Model Kairouz-Wahbe et al. (2008) found that Bit1 (Ptrh2)-null mice were born alive, but they died within the first 2 weeks from a runting syndrome with muscle weakness, ataxia, and decreased weight.
Complications [ edit ] Early complications include shock , infection, systemic inflammatory response syndrome , low blood calcium, high blood glucose, and dehydration . ... Severe inflammation can lead to intra-abdominal hypertension and abdominal compartment syndrome , further impairing renal and respiratory function and potentially requiring management with an open abdomen to relieve the pressure. [9] Late complications include recurrent pancreatitis and the development of pancreatic pseudocysts —collections of pancreatic secretions that have been walled off by scar tissue. ... The Modified Glasgow criteria suggests that a case be considered severe if at least three of the following are true: [38] Age > 55 years Blood levels: PO2 oxygen < 60 mmHg or 7.9 kPa White blood cells > 15,000/µlitre Calcium < 2 mmol/litre Blood urea nitrogen > 16 mmol/litre Lactate dehydrogenase (LDH) > 600iu/litre Aspartate transaminase (AST) > 200iu/litre Albumin < 32g/litre Glucose > 10 mmol/litre This can be remembered using the mnemonic PANCREAS: PO2 oxygen < 60 mmHg or 7.9 kPa Age > 55 Neutrophilia white blood cells > 15,000/µlitre Calcium < 2 mmol/litre Renal function ( BUN ) > 16 mmol/litre Enzymes lactate dehydrogenase (LDH) > 600iu/litre aspartate transaminase (AST) > 200iu/litre Albumin < 32g/litre Sugar glucose > 10 mmol/litre The BISAP score ( b lood urea nitrogen level >25 mg/dl (8.9 mmol/L), i mpaired mental status, s ystemic inflammatory response syndrome , a ge over 60 years, p leural effusion) has been validated as similar to other prognostic scoring systems. [39] Epidemiology [ edit ] Globally the incidence of acute pancreatitis is 5 to 35 cases per 100,000 people. ... National Library of Medicine. v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum v t e Alcohol and health Alcohol use Alcohol-related crimes Drunk drivers Alcohol-related traffic crashes in the United States Driving under the influence (DUI) Drunk driving in the United States Public intoxication Rum-running Adulterated moonshine / Denatured alcohol List of methanol poisoning incidents Alcoholism Alcohol and Native Americans Alcoholism in adolescence Alcoholism in family systems Collaborative Study on the Genetics of Alcoholism College student alcoholism Disease theory of alcoholism High-functioning alcoholic (HFA) Seeing pink elephants Chemistry Beer chemistry Congener Alcohol congener analysis Ethanol Blood alcohol content Breathalyzer Fusel alcohol Wine chemistry Effects Short-term effects of alcohol consumption Long-term effects of alcohol On memory Subjective response to alcohol Interactions Aging Brain Cancer breast cancer Cortisol Pregnancy Sleep Tolerance / intolerance Weight Beverage-specific Beer: Potomania Red wine: Red wine headache Social issues Alcohol advertising on college campuses Sex Alcohol myopia Alcohol abuse among college students Binge drinking Epidemiology Blackout (alcohol-related amnesia) Blackout Wednesday Drinking game list pregaming Drinking in public Drunk dialing Drunk walking Drunkorexia Dry drunk French paradox Hair of the dog Nightcap Pantsdrunk Passive drinking Binge drinking devices Beer bong Yard of ale Routes of administration Alcohol enema Alcohol inhalation Sconcing Surrogate alcohol Related issues Balconing Suicide History Dionysian Mysteries Dipsomania Gin Craze List of deaths through alcohol Rum ration Speakeasy General Beer day Drinking culture Apéritif and digestif Hangover remedies Health effects of wine Wine and food matching Long-distance race involving alcohol List of countries by alcohol consumption per capita Alcohol consumption by youth in the United States Nip joint Alcohol control Alcohol law Administrative license suspension (ALS) Alcohol packaging warning messages Drunk driving law by country DWI court Field sobriety testing Hip flask defence Ignition interlock device Legal drinking age Age controversy in US Underage drinking in US List of alcohol laws of US Alcohol prohibition List of countries with alcohol prohibition Neo-prohibitionism Temperance movement Sobriety Alcohol detoxification Alcohol-free zone Dry campus United States open-container laws Designated driver Alcohol rehabilitation Drunk tank Managed alcohol program Non-alcoholic drink List of cocktails List of mixed drinks Spritzer Malt drinks Teetotalism Temperance bar Twelve-step groups Al-Anon/Alateen Alcoholics Anonymous (AA): Adult Children of Alcoholics (ACA) Alcohol limitation 0-0-1-3 Alcohol education Alcohol server training FRAMES Dry January Foundation for Advancing Alcohol Responsibility Campaigns Get Your Sexy Back Liquor license Low-alcohol drinks Fermented tea Low-alcohol beer Low-alcoholic malt drinks Small beer Measurement Alcoholic spirits measure Standard drink Recommended maximum intake of alcoholic beverages Addiction medicine Disulfiram-like drugs : disulfiram , calcium carbimide , cyanamide . Sulfonic acids : Acamprosate Religion and alcohol Christian views on alcohol alcohol in the Bible Islam and alcohol History Bratt System Related Index of alcohol-related articles Austrian syndrome Ban on caffeinated alcoholic beverages Brief intervention Gateway drug effect Last call Mood disorder Non-alcoholic fatty liver disease Self-medication Spins Sober companion Sober living houses Sobering center Town drunk Category Authority control GND : 4004723-4 NDL : 00571544
Overview Pancreatitis is inflammation of the pancreas. The pancreas is a long, flat gland that sits tucked behind the stomach in the upper abdomen. The pancreas produces enzymes that help digestion and hormones that help regulate the way your body processes sugar (glucose). Pancreatitis can occur as acute pancreatitis — meaning it appears suddenly and lasts for days. Some people develop chronic pancreatitis, which is pancreatitis that occurs over many years. Mild cases of pancreatitis improve with treatment, but severe cases can cause life-threatening complications.
Silencing of the FMR1 gene in Fragile X syndrome . FMR1 co-localizes with a rare fragile site, visible here as a gap on the long arms of the X chromosome . ... Clinically, the most important rare fragile site is FRAXA, which is associated with the fragile X syndrome , the most common cause of hereditary intellectual disability. ... The FRAXA site is perhaps most famous for its role in Fragile X syndrome , but fragile sites are clinically implicated in many other important diseases, such as cancer . ... "Association of a chromosome deletion syndrome with a fragile site within the proto-oncogene CBL2". ... "Chromosomal instability at common fragile sites in Seckel syndrome" . American Journal of Human Genetics . 75 (4): 654–60. doi : 10.1086/422701 .
Overview Impetigo (im-puh-TIE-go) is a common and highly contagious skin infection that mainly affects infants and young children. It usually appears as reddish sores on the face, especially around the nose and mouth and on the hands and feet. Over about a week, the sores burst and develop honey-colored crusts. Impetigo Impetigo starts as a reddish sore that ruptures, oozes for a few days and then forms a honey-colored crust. Sores mainly occur around the nose and mouth in infants and children. Treatment with antibiotics can limit the spread of impetigo to others.
Nutritional [ edit ] Though not always grouped together with the inherited versions, a severe nutritional deficiency of vitamin B 12 can also result in syndrome with identical symptoms and treatments as the genetic methylmalonic acidemias. [10] Methylmalonyl CoA requires vitamin B 12 to form succinyl-CoA. ... Like the mutase, the epimerase also functions in breaking down the same substances, but to a significantly lesser extent than the mutase does. [8] The phenotypic differences caused by a deficiency of the epimerase as opposed to the mutase are so mild that there is debate within the medical community as to whether or not this genetic deficiency can be considered a disorder or clinical syndrome. [13] Adenosylcobalamin [ edit ] Also known as vitamin B 12, this form of cobalamin is a required cofactor of methylmalonyl CoA mutase. ... PMID 17075691 . ^ Higginbottom MC, Sweetman L, Nyhan WL (1978). "A syndrome of methylmalonic aciduria, homocystinuria, megaloblastic anemia and neurological abnormalities in a vitamin B 12 -deficient breast-fed infant of a strict vegetarian". ... Further reading [ edit ] GeneReviews article on Methylmalonic Acidemia GeneReviews article on Disorders of Intracellular Cobalamin Metabolism External links [ edit ] Classification D ICD - 10 : E71.1 ICD - 10-CM : E71.120 ICD - 9-CM : 270.3 OMIM : 251000 251100 251110 277380 277400 277410 606169 DiseasesDB : 29509 External resources MedlinePlus : 001162 eMedicine : neuro/576 v t e Inborn error of amino acid metabolism K → acetyl-CoA Lysine /straight chain Glutaric acidemia type 1 type 2 Hyperlysinemia Pipecolic acidemia Saccharopinuria Leucine 3-hydroxy-3-methylglutaryl-CoA lyase deficiency 3-Methylcrotonyl-CoA carboxylase deficiency 3-Methylglutaconic aciduria 1 Isovaleric acidemia Maple syrup urine disease Tryptophan Hypertryptophanemia G G→ pyruvate → citrate Glycine D-Glyceric acidemia Glutathione synthetase deficiency Sarcosinemia Glycine → Creatine : GAMT deficiency Glycine encephalopathy G→ glutamate → α-ketoglutarate Histidine Carnosinemia Histidinemia Urocanic aciduria Proline Hyperprolinemia Prolidase deficiency Glutamate / glutamine SSADHD G→ propionyl-CoA → succinyl-CoA Valine Hypervalinemia Isobutyryl-CoA dehydrogenase deficiency Maple syrup urine disease Isoleucine 2-Methylbutyryl-CoA dehydrogenase deficiency Beta-ketothiolase deficiency Maple syrup urine disease Methionine Cystathioninuria Homocystinuria Hypermethioninemia General BC / OA Methylmalonic acidemia Methylmalonyl-CoA mutase deficiency Propionic acidemia G→ fumarate Phenylalanine / tyrosine Phenylketonuria 6-Pyruvoyltetrahydropterin synthase deficiency Tetrahydrobiopterin deficiency Tyrosinemia Alkaptonuria / Ochronosis Tyrosinemia type I Tyrosinemia type II Tyrosinemia type III / Hawkinsinuria Tyrosine → Melanin Albinism : Ocular albinism ( 1 ) Oculocutaneous albinism ( Hermansky–Pudlak syndrome ) Waardenburg syndrome Tyrosine → Norepinephrine Dopamine beta hydroxylase deficiency reverse: Brunner syndrome G→ oxaloacetate Urea cycle / Hyperammonemia ( arginine aspartate ) Argininemia Argininosuccinic aciduria Carbamoyl phosphate synthetase I deficiency Citrullinemia N-Acetylglutamate synthase deficiency Ornithine transcarbamylase deficiency / translocase deficiency Transport / IE of RTT Solute carrier family : Cystinuria Hartnup disease Iminoglycinuria Lysinuric protein intolerance Fanconi syndrome : Oculocerebrorenal syndrome Cystinosis Other 2-Hydroxyglutaric aciduria Aminoacylase 1 deficiency Ethylmalonic encephalopathy Fumarase deficiency Trimethylaminuria v t e Metabolic disorders of vitamins , coenzymes, and cofactors B7 Biotin / MCD Biotinidase deficiency Holocarboxylase synthetase deficiency Other B B5 ( Pantothenate kinase-associated neurodegeneration ) B12 ( Methylmalonic acidemia ) Other vitamin Familial isolated vitamin E deficiency Nonvitamin cofactor Tetrahydrobiopterin deficiency Molybdenum cofactor deficiency
Maternal B 12 deficiency can produce a methylmalonic acidemia syndrome in an infant that ranges from severe encephalopathy to elevated serum concentration of propionylcarnitine (C3) detected by newborn screening [Chace et al 2001]. ... (See SUCLG1 -Related Mitochondrial DNA Depletion Syndrome, Encephalomyopathic Form with Methylmalonic Aciduria.) Biallelic SUCLA2 pathogenic variants are associated with hypotonia, muscle atrophy presenting around ages three to six months (with mtDNA depletion, complex I, III, and IV deficiency in the muscle), hyperkinesia, seizures, severe hearing impairment, and growth failure. Patients develop a Leigh syndrome-like disorder, cortical and basal ganglia atrophy, and dystonia. ... (See SUCLA2 -Related mtDNA Depletion Syndrome, Encephalomyopathic Form with Methylmalonic Aciduria.) Methylmalonic aciduria ranges from 10 to 200 mmol/mol creatinine in these individuals and is accompanied by raised plasma concentrations of lactate, methylcitrate, 3-hydroxyproprionic and 3-hydroxyisovaleric acid, proprionylcarnitine, and C4-dicarboxylic carnitine (C4DC) [Elpeleg et al 2005, Carrozzo et al 2007, Ostergaard et al 2007, Morava et al 2009]. Reye-like syndrome. A Reye-like syndrome of hepatomegaly and obtundation in the face of a mild intercurrent infection can be seen as an unrecognized presentation of a number of inborn errors of metabolism, including isolated methylmalonic acidemia [Chang et al 2000].
Methylmalonic acidemia refers to a group of inherited conditions in which the body can’t breakdown certain parts of proteins and fats. This leads to a build-up of toxic substances and bouts of serious illness called decompensation events or metabolic crises. Symptoms of a decompensation event include poor feeding, vomiting, trouble breathing, and lack of energy (lethargy). These can occur at different ages and can range from mild to severe. Methylmalonic acidemia is caused by changes in several different genes and is inherited in an autosomal recessive fashion. Treatment includes aggressive management of decompensation events, a low-protein diet, certain medications, antibiotics and, in some cases, liver and kidney transplantation.
Some individuals with FLNA -related PVNH display connective tissue and vascular anomalies also seen in classic Ehlers-Danlos syndrome [Sheen et al 2005]. In a recent review of the vascular and connective tissue anomalies associated with pathogenic variants in FLNA , ten of the eleven affected individuals showed one or more congenital cardiac or vascular anomalies. ... Note: Although three affected brothers with West syndrome/hypsarrhythmia were initially reported to have a hemizygous FLNA pathogenic variant [Masruha et al 2006] this reported sequence variant has subsequently been thought to be a rare polymorphism [Robertson 2006]. ... Periventricular nodular heterotopia also occurs in the following syndromes (whether each of these represents a truly distinct disorder or FLNA -related PVNH plus a concurrent condition remains to be determined): Nonfamilial PVNH caused by perinatal insult or chromosomal rearrangement Autosomal recessive PVNH (OMIM 608097). ... Autosomal dominant forms of PVNH (OMIM 608098) (chromosome 5p15, 1p36, 7q11) [Sheen et al 2003b, Neal et al 2006, Ferland et al 2009] Bilateral periventricular nodular heterotopia (BPNH)/frontonasal malformations (OMIM 300049) [Guerrini & Dobyns 1998] PVNH (unilateral/bilateral and isolated) in two boys with fragile X syndrome [Moro et al 2006] BPNH with micronodules BPNH with ambiguous genitalia BPNH with microcephaly BPNH/intellectual disability/syndactyly [Dobyns et al 1997] BPNH/nephrosis syndrome BPNH/short gut syndrome Unilateral PVNH Bilateral anterior PVNH with fronto-perisylvian polymicrogyria [Parrini et al 2006] Bilateral PVNH involving temporo-occipital and trigones with hippocampal malformation, and subclassified into polymicrogyria or cerebellar hypoplasia or hydrocephalus [Parrini et al 2006] Periventricular nodular heterotopia, intellectual disability, and epilepsy associated with 5q14.3-q15 deletion (OMIM 612881) [Cardoso et al 2009] Laminar heterotopia occurring in deep white matter and band-like heterotopia occurring between the cortex and ventricular surface are seen in X-linked subcortical band heterotopia. ... Currently no guidelines exist on the most appropriate surveillance for and management of cardiac, vascular, and connective tissue problems during pregnancy in women with PVNH. See Marfan Syndrome and Ehlers-Danlos Syndrome, Classic Type for possible pregnancy management recommendations.
Some people also have hyperflexible joints and vascular anomalies, which also occur in Ehlers-Danlos syndrome (EDS). X-linked periventricular heterotopia is caused by mutations in the FLNA gene and is inherited in an X-linked dominant manner.
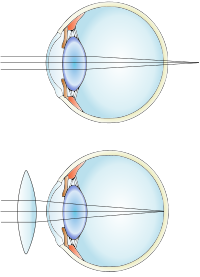
Overview Farsightedness (hyperopia) is a common vision condition in which you can see distant objects clearly, but objects nearby may be blurry. The degree of your farsightedness influences your focusing ability. People with severe farsightedness may be able to clearly see only objects a great distance away, while those with mild farsightedness may be able to clearly see objects that are closer. Farsightedness usually is present at birth and tends to run in families. You can easily correct this condition with eyeglasses or contact lenses. Another treatment option is surgery. Symptoms Farsightedness may mean: Nearby objects may appear blurry You need to squint to see clearly You have eyestrain, including burning eyes, and aching in or around the eyes You have general eye discomfort or a headache after doing close tasks, such as reading, writing, computer work or drawing, for a time When to see a doctor If your degree of farsightedness is pronounced enough that you can't perform a task as well as you wish, or if your quality of vision detracts from your enjoyment of activities, see an eye doctor.
King et al. (1984) described 2 cases of what they called trichothiodystrophy-neurotrichocutaneous syndrome of Pollitt in unrelated children. ... King et al. (1984) suggested that this disorder is the same as the Amish brittle hair syndrome (234050) and the Sabinas brittle hair syndrome (211390). Happle et al. (1984) reported a patient with congenital ichthyosis with trichothiodystrophy (Tay syndrome) and reviewed 12 previously reported patients. ... In the son of a Finnish uncle-niece marriage, Blomquist et al. (1991) observed Tay or IBIDS syndrome, which was manifested by growth and mental retardation, congenital ichthyosis, and brittle hair. ... Kleijer et al. (1994) described a female child with what Crovato et al. (1983) and Rebora and Crovato (1988) referred to as the PIBI(D)S syndrome with trichothiodystrophy. She had photosensitivity, ichthyosis, brittle hair, impaired intelligence, possibly decreased fertility, and short stature.
In extreme cases the blood flow may be so restricted that the left ventricle fails to grow, resulting in hypoplastic left heart syndrome (241550) (Garg et al., 2005). The valve calcification often observed in bicuspid aortic valve is a result of inappropriate activation of osteoblast-specific gene expression. ... The pathology of the aortic media was very similar to that found in Marfan syndrome. Burks et al. (1998) recommended echocardiographic surveillance of the ascending aorta at regular intervals and consideration of beta-adrenergic blockade in patients with significant dilation. ... Two families had nonmanifesting obligate carriers, and 3 families had additional left outflow tract anomalies including coarctation of the aorta in 2 families and hypoplastic left heart syndrome in 1 family, suggesting reduced penetrance and variable expressivity. ... McBride et al. (2008) analyzed the NOTCH1 gene in 91 unrelated European American patients with congenital aortic valve stenosis, bicuspid aortic valve, coarctation of the aorta (COA; see 120000), and/or hypoplastic left heart syndrome (HLHS; see 241550), and identified heterozygous missense variants in 6 probands. ... The male preponderance for bicuspid aortic valve is of interest in relation to the fact that this anomaly is frequent in the XO Turner syndrome where it may be the most common cardiac defect; Miller et al. (1983) found that 12 of 35 consecutive patients with Turner syndrome (34%) had isolated, nonstenotic bicuspid aortic valve, as demonstrated by echocardiography.
Familial bicuspid aortic valve is a rare, genetic, aortic malformation defined as a presence of abnormal two-leaflet aortic valve in at least 2 first-degree relatives. It is frequently asymptomatic or may be associated with progressive aortic valve disease (aortic regurgitation and/or aortic stenosis, typically due to valve calcification) and a concomitant aortopathy (i.e. aortic dilation, aortic aneurysm and/or dissection).
A number sign (#) is used with this entry because of evidence that aortic valve disease-2 (AOVD2) is caused by heterozygous mutation in the SMAD6 gene (602931) on chromosome 15q22. Description Aortic valve disease-2 (AOVD2) is characterized by bicuspid aortic valve (BAV) and dilation of the ascending aorta. Calcification of the valve and the aorta has been observed, and some patients exhibit coarctation of the aorta (Tan et al., 2012; Luyckx et al., 2019; Park et al., 2019). For a general phenotypic description and a discussion of genetic heterogeneity of aortic valve disease, see AOVD1 (109730). Clinical Features Tan et al. (2012) studied 2 patients with aortic valve disease.
Hypothyroidism Diffuse and lobulated [7] Thyroid hormone replacement Prevalence: 1 to 1.5 in a 1000 Remission with treatment Pituitary disease Hypersecretion of thyroid stimulating hormone , almost always by a pituitary adenoma [8] Diffuse Pituitary surgery Very rare [8] Graves' disease —also called Basedow syndrome Autoantibodies (TSHR-Ab) that activate the TSH -receptor (TSHR) Hyperthyroidism Diffuse Antithyroid agents , radioiodine , surgery Will develop in about 0.5% of males and 3% of females Remission with treatment, but still lower quality of life for 14 to 21 years after treatment, with lower mood and lower vitality, regardless of the choice of treatment [9] Thyroiditis Acute or chronic inflammation Can be hyperthyroidism initially, but progress to hypothyroidism Thyroid cancer Usually uninodular Overall relative 5-year survival rate of 85% for females and 74% for males [10] Benign thyroid neoplasms Usually hyperthyroidism Usually uninodular Mostly harmless [11] Thyroid hormone insensitivity Secretional hyperthyroidism , Symptomatic hypothyroidism Diffuse Sarcoidosis Amyloidosis Hydatidiform mole Cysts Acromegaly Pendred syndrome Diagnosis [ edit ] Goitre with toxic adenoma Goitre may be diagnosed via a thyroid function test in an individual suspected of having it. [12] Types [ edit ] A goitre may be classified either as nodular or diffuse. ... ISBN 0-671-62028-2 . ^ Basedow's syndrome or disease at Who Named It? – the history and naming of the disease ^ Ljunggren JG (August 1983). "[Who was the man behind the syndrome: Ismail al-Jurjani, Testa, Flagani, Parry, Graves or Basedow? ... External links [ edit ] Classification D ICD - 10 : E01 - E05 ICD - 9-CM : 240.9 MeSH : D006042 DiseasesDB : 5332 External resources MedlinePlus : 001178 Wikimedia Commons has media related to Goiters . v t e Thyroid disease Hypothyroidism Iodine deficiency Cretinism Congenital hypothyroidism Myxedema Myxedema coma Euthyroid sick syndrome Signs and symptoms Queen Anne's sign Woltman sign Thyroid dyshormonogenesis Pickardt syndrome Hyperthyroidism Hyperthyroxinemia Thyroid hormone resistance Familial dysalbuminemic hyperthyroxinemia Hashitoxicosis Thyrotoxicosis factitia Thyroid storm Graves' disease Signs and symptoms Abadie's sign of exophthalmic goiter Boston's sign Dalrymple's sign Stellwag's sign lid lag Griffith's sign Möbius sign Pretibial myxedema Graves' ophthalmopathy Thyroiditis Acute infectious Subacute De Quervain's Subacute lymphocytic Palpation Autoimmune /chronic Hashimoto's Postpartum Riedel's Enlargement Goitre Endemic goitre Toxic nodular goitre Toxic multinodular goiter Thyroid nodule Colloid nodule
Overview A goiter (GOI-tur) is the irregular growth of the thyroid gland. The thyroid is a butterfly-shaped gland located at the base of the neck just below the Adam's apple. A goiter may be an overall enlargement of the thyroid, or it may be the result of irregular cell growth that forms one or more lumps (nodules) in the thyroid. A goiter may be associated with no change in thyroid function or with an increase or decrease in thyroid hormones. Enlarged thyroid Widespread enlargement of the thyroid can expand the gland well beyond its typical size (left) and cause a noticeable bulge in the neck (right).