Fragile X syndrome is a genetic condition that causes a range of developmental problems including learning disabilities and cognitive impairment. ... Children with fragile X syndrome may also have anxiety and hyperactive behavior such as fidgeting or impulsive actions. ... Seizures occur in about 15 percent of males and about 5 percent of females with fragile X syndrome. Most males and about half of females with fragile X syndrome have characteristic physical features that become more apparent with age. ... Causes Mutations in the FMR1 gene cause fragile X syndrome. The FMR1 gene provides instructions for making a protein called FMRP. ... The premutation is also associated with an increased risk of disorders called fragile X-associated primary ovarian insufficiency (FXPOI) and fragile X-associated tremor/ataxia syndrome (FXTAS). Learn more about the gene associated with Fragile X syndrome FMR1 Inheritance Pattern Fragile X syndrome is inherited in an X-linked dominant pattern .
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a rare neurodegenerative disorder characterized by adult-onset progressive intention tremor and gait ataxia.
Fragile X–Associated Tremor/Ataxia Syndrome. Archives of Neurology. VOL 65 (NO. 1), Jan 2008 ^ Jump up to: a b ^ Jump up to: a b c Jump up^ ^ a b c "Fragile X-Associated Tremor and Ataxia Syndrome (FXTAS): Overview" . www.nichd.nih.gov . ... PMID 28814538 . ^ The fragile X-associated tremor ataxia syndrome (FXTAS) . Springer New York. 2010. doi : 10.1007/978-1-4419-5805-1 . ... "Fragile X-Associated Tremor/Ataxia Syndrome: From Molecular Pathogenesis to Development of Therapeutics" . ... "Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome" . Science . 343 (6174): 1002–1005. ... "Fragile X-Associated Tremor Ataxia Syndrome: The Expanding Clinical Picture, Pathophysiology, Epidemiology, and Update on Treatment" .
Fragile X-associated tremor/ataxia syndrome (FXTAS) is characterized by problems with movement and thinking ability (cognition). ... An expansion of more than 200 repeats, a full mutation, causes a more serious condition called fragile X syndrome, which is characterized by intellectual disability, learning problems, and certain physical features. ... However, it may cause mild versions of the features seen in fragile X syndrome, such as prominent ears, anxiety, and mood swings. Learn more about the gene associated with Fragile X-associated tremor/ataxia syndrome FMR1 Inheritance Pattern An increased risk of developing FXTAS is inherited in an X-linked dominant pattern .
A number sign (#) is used with this entry because fragile X tremor/ataxia syndrome (FXTAS) is caused by an expanded trinucleotide repeat in the FMR1 gene (309550.0004). ... Description Jacquemont et al. (2007) provided a review of fragile X syndrome, which they characterized as a neurodevelopmental disorder, and FXTAS, which they characterized as a neurodegenerative disorder. Amiri et al. (2008) provided a review of FXTAS and noted that the pathogenesis of the disorder is distinct from that in fragile X syndrome. FXTAS results form a toxic gain of function of FMR1 RNA, whereas fragile X syndrome results from a loss of FMR1 function. ... Fourteen male carriers with FXTAS syndrome and 14 age- and education-matched controls were assessed with the Neuropsychiatric Inventory (NPI) profile, formal cognitive testing, and genetic analysis. ... His grandson had full fragile X mental retardation syndrome. Goncalves et al. (2007) emphasized the unusual presentation of FXTAS in the elder patient.
Miller syndrome Other names Mandibulfacial dysostosis with postaxial limb anomalies This condition is inherited in an autosomal recessive manner. Miller syndrome , also known as Genée–Wiedemann syndrome , Wildervanck–Smith syndrome or postaxial acrofacial dystosis , is an extremely rare genetic condition that manifests as craniofacial, limb and eye deformities. ... Additional features of the syndrome include downward-slanting palpebral fissures, malar hypoplasia , malformed ears , and a broad nasal ridge. ... "Postaxial acrofacial dysostosis syndrome". The Journal of Pediatrics . 95 (6): 970–5. doi : 10.1016/S0022-3476(79)80285-1 . PMID 501501 . ^ Opitz JM, Stickler GB (August 1987). "The Genée-Wiedemann syndrome, an acrofacial dysostosis--further observation".
Miller syndrome is a rare condition that mainly affects the development of the face and limbs. ... Individuals with Miller syndrome have various bone abnormalities in their arms and legs. ... Causes Mutations in the DHODH gene cause Miller syndrome. This gene provides instructions for making an enzyme called dihydroorotate dehydrogenase. ... It remains unclear exactly how DHODH gene mutations lead to abnormal development of the pharyngeal arches in people with Miller syndrome. Development of the arms and legs is also affected by Miller syndrome. ... It is also unknown how mutations in the DHODH gene cause bone abnormalities in the arms and legs of people with Miller syndrome. Learn more about the gene associated with Miller syndrome DHODH Inheritance Pattern This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations.
Miller syndrome is a rare condition that mainly affects the development of the face and limbs. ... The severity of the disorder varies among affected individuals. Miller syndrome is caused by mutations in the DHODH gene.
Vigneron et al. (1991) reported a case and suggested that the mandibulofacial dysostosis is similar to that of Treacher Collins syndrome (154500). The postaxial deficiency in the limbs distinguishes the disorder from Nager syndrome (154400), which has preaxial limb deficiency. ... Pereira et al. (1992) tabulated the main clinical features in sporadic and familial cases of this syndrome, which they referred to as the Genee-Wiedemann syndrome (Genee, 1969; Wiedemann, 1973). ... Sanger sequencing confirmed the presence of DHODH mutations in 3 additional families with Miller syndrome. Collectively, Ng et al. (2010) identified 11 different mutations in the DHODH gene in 6 kindreds with Miller syndrome by a combination of exome and targeted resequencing. ... One mutation (126064.0001) was shared by 2 unrelated individuals with Miller syndrome who were of different self-identified geographic ancestry. ... This is the same pathway affected by mutations in DHODH, which result in Miller syndrome (Ng et al., 2010). History Because of phenotypic similarities between Miller syndrome and Treacher Collins syndrome (154500), which is caused by mutations in the TCOF1 gene (606847), Splendore et al. (2002) screened the TCOF1 gene in 1 Miller syndrome patient, but found no mutations.
Differential diagnosis The clinical phenotype of Miller syndrome overlaps with mandibulofacial and other acrofacial dysostosis syndromes including Treacher Collins, Guion-Almeida (mandibulofacial dysostosis with microcephaly) and Nager syndromes.
Bruck syndrome Other names Osteogenesis imperfecta-congenital joint contractures syndrome Bruck syndrome is inherited in an autosomal recessive manner Specialty Rheumatology Bruck syndrome is characterized as the combination of arthrogryposis multiplex congenita and osteogenesis imperfecta . Both diseases are uncommon, but concurrence is extremely rare which makes Bruck syndrome very difficult to research. [1] Bruck syndrome is thought to be an atypical variant of osteogenesis imperfecta most resembling type III, if not its own disease. [2] [3] Multiple gene mutations associated with osteogenesis imperfecta are not seen in Bruck syndrome. ... Bruck syndrome type 1 is caused by a homozygous mutation in the FKBP10 gene. ... "Osteogenesis imperfecta with joint contractures: Bruck syndrome". Pediatric Radiology . 28 (2): 117–119. doi : 10.1007/s002470050309 . ... "Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans".
Kuskokwim syndrome is characterized by joint deformities called contractures that restrict the movement of affected joints. ... Affected individuals are typically shorter than their peers and they may have an abnormally large head (macrocephaly ). Frequency Kuskokwim syndrome is extremely rare. It affects a small number of people from the Yup'ik Eskimo population in southwest Alaska. Causes Kuskokwim syndrome is caused by mutations in the FKBP10 gene, which provides instructions for making the FKBP10 protein (formerly known as FKBP65). ... As a result, people with Kuskokwim syndrome have only about 5 percent of the normal amount of FKBP10 protein. ... It is unclear how these changes in the collagen matrix are involved in the development of joint contractures and other abnormalities in people with Kuskokwim syndrome. Learn more about the gene associated with Kuskokwim syndrome FKBP10 Inheritance Pattern This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations.
Not to be confused with Meige's syndrome or Meige lymphedema . Meigs's syndrome Specialty Gynecologic oncology In medicine , Meigs's syndrome , also Meigs syndrome or Demons–Meigs syndrome , is the triad of ascites , pleural effusion , and benign ovarian tumor ( ovarian fibroma , fibrothecoma, Brenner tumour , and occasionally granulosa cell tumour ). [1] [2] [3] Meigs syndrome resolves after the resection of the tumor . ... Meigs syndrome is characterized by the presence of a benign solid ovarian tumor associated with ascites and right hydrothorax that disappear after tumor removal. ... Eponym [ edit ] Meigs syndrome is named for Joe Vincent Meigs . [6] [7] [8] References [ edit ] ^ a b c Meigs syndrome at eMedicine ^ Morán-Mendoza A, Alvarado-Luna G, Calderillo-Ruiz G, Serrano-Olvera A, López-Graniel CM, Gallardo-Rincón D (2006). "Elevated CA125 level associated with Meigs' syndrome: case report and review of the literature". ... PMC 5008483 . PMID 26656338 . ^ Meigs' syndrome at Who Named It? ^ Lurie S (October 2000).
Meigs syndrome is a rare neoplastic disease characterized by the clinical triad of benign ovarian tumor (typically, ovarian fibroma or fibroma-like tumor), hydrothorax and ascites, which resolve after tumor resection.
A number sign (#) is used with this entry because of evidence that congenital amegakaryocytic thrombocytopenia (CAMT) can be caused by homozygous or compound heterozygous mutation in the myeloproliferative leukemia virus oncogene (MPL; 159530) on chromosome 1p34. Description Congenital amegakaryocytic thrombocytopenia (CAMT) is a rare disorder expressed in infancy and characterized by isolated thrombocytopenia and megakaryocytopenia with no physical anomalies (Muraoka et al., 1997). King et al. (2005) proposed a new classification of CAMT based on the course and outcome of the disease, as exemplified by 20 patients: CAMT type I (11 patients) was characterized by early onset of severe pancytopenia, decreased bone marrow activity, and very low platelet counts. CAMT type II (9 patients) was somewhat milder and characterized by transient increases of platelet counts up to nearly normal values during the first year of life and an onset of bone marrow failure at age 3 or later. Clinical Features Muraoka et al. (1997) found that a patient with CAMT had a defective response to thrombopoietin (TPO; 600044) in megakaryocyte-colony formation, decreased numbers of erythroid and myelocytic progenitors in clonal cultures, a lack of MPL mRNA in bone marrow mononuclear cells, and an elevated serum level of TPO.
Clinical Features Stickler Syndrome, Type I, Predominantly Ocular Individuals with Stickler syndrome caused by mutations in the COL2A1 gene (Sticker syndrome type I, or STL1; 108300) almost always display a congenital vitreous abnormality consisting of a vestigial gel in the retrolental space, bounded by a highly folded membrane. In a study of 50 families presenting with the Stickler syndrome type I membranous vitreous phenotype, Richards et al. (2006) identified 3 families with a predominantly ocular form of type I Stickler syndrome in which 3 distinct mutations in the alternatively spliced exon 2 of the COL2A1 gene segregated with the respective phenotypes. ... Wagner syndrome is often confused with Stickler syndrome. Like the predominantly ocular or ocular-only phenotype caused by certain mutations in COL2A1 (Richards et al., 2006), Wagner syndrome has no systemic features. However, the vitreoretinal phenotype is different, as neither of the recognized vitreous abnormalities in Stickler syndrome are present in Wagner syndrome and there is a lower incidence of retinal detachment. ... Molecular Genetics Stickler Syndrome, Type I, Predominantly Ocular In 3 families with a predominantly ocular form of type I Stickler syndrome, Richards et al. (2006) found 3 distinct mutations in the alternatively spliced exon 2 of the COL2A1 gene that segregated with the phenotype.
A rare, hereditary, non-syndromic form of vitreoretinopathy characterized by retinal tears due to abnormal vitreous, and commonly present refractive errors. No other signs or symptoms of Stickler syndrome is present.
A number sign (#) is used with this entry because of evidence that this X-linked form of infantile X-linked spinal muscular atrophy (SMAX2) is caused by mutations in the UBE1 gene (314370) at Xp11. Description X-linked infantile spinal muscular atrophy (XL-SMA) is characterized by neonatal onset of severe hypotonia, areflexia, and multiple congenital contractures, known as arthrogryposis, associated with loss of anterior horn cells and infantile death (summary by Ramser et al., 2008). Historically, Hall et al. (1982) distinguished at least 3 clinical varieties of X-linked arthrogryposis. (1) One family had a severe lethal form with severe contractures, scoliosis, chest deformities, hypotonia, micrognathia, and death from respiratory insufficiency by age 3 months. Apparently progressive loss of anterior horn cells was the cause. (2) Two families had moderately severe AMC associated with ptosis, microphallus, cryptorchidism, inguinal hernias, and normal intelligence. Nonprogressive intrauterine myopathy appeared to be the 'cause'. (3) In 2 families and a sporadic case, the disorder took the form of a resolving AMC, with mild to moderate contractures improving dramatically with time, normal intelligence, and no other anomalies; tight connective tissues on misplaced tendons was postulated.
X-linked infantile spinal muscular atrophy is a condition that affects only boys and is characterized by severe muscle weakness and absent reflexes (areflexia). Affected children often have multiple joint deformities (contractures) from birth that cause joint stiffness (arthrogryposis) and impair movement. In severe cases, affected infants are born with broken bones. The muscle weakness worsens over time; affected children reach some early motor developmental milestones, such as sitting unassisted, but these skills are often lost (developmental regression). Additional features of X-linked infantile spinal muscular atrophy include an unusually small chin (micrognathia ), abnormal curvature of the spine (scoliosis or kyphosis ), and undescended testes (cryptorchidism). Weakness of the chest muscles used for breathing often leads to life-threatening breathing problems.
X-linked distal arthrogryposis multiplex congenital (SMAX2) is a rare form of spinal muscular atrophy characterized by the neonatal onset of severe hypotonia, areflexia, profound weakness, multiple congenital contractures, facial dysmorphic features (myopathic face with open, tent-shaped mouth), cryptorchidism, and mild skeletal abnormalities (i.e. kyphosis, scoliosis), that is often preceded by polyhydramnios and reduced fetal movements in utero and followed by bone fractures shortly after birth. SMAX2 patients often have a limited life span, often succumbing to the disease within 2 years, as muscle weakness is progressive and chest muscle involvement eventually leads to ventilatory insufficiency and respiratory failure.
A number sign (#) is used with this entry because of evidence that ichthyosis vulgaris is caused by heterozygous mutation in the filaggrin gene (FLG; 135940) on chromosome 1q21. Patients with homozygous or compound heterozygous mutations in this gene have a more severe phenotype. Clinical Features Ichthyosis is one of the most frequent single-gene disorders in humans. The most widely cited incidence figure is 1 in 250 based on a survey of 6,051 healthy English schoolchildren (Wells and Kerr, 1966). The phenotypic characteristics of ichthyosis vulgaris include palmar hyperlinearity, keratosis pilaris, and a fine scale that is most prominent over the lower abdomen, arms, and legs.
Ichthyosis vulgaris is a common skin disorder passed down through families that leads to dry, scaly skin. It often begins in early childhood. Treatment may include heavy duty moisturizers which contain chemicals that help the skin to shed normally, including lactic acid , salicylic acid, and urea. Ichthyosis vulgaris can be a nuisance, but it rarely affects overall health. The condition usually disappears during adulthood, but may return in later years. This condition is inherited in an autosomal dominant pattern.
Plummer-Vinson or Paterson-Kelly syndrome presents as a classical triad of dysphagia, iron-deficiency anemia and esophageal webs. Epidemiology Exact data about the epidemiology of the syndrome are not available; the syndrome is extremely rare. ... One of the most important clinical aspects of Plummer-Vinson syndrome is the association with upper alimentary tract cancers. Etiology The etiopathogenesis of Plummer-Vinson syndrome is unknown. The most important possible etiological factor is iron deficiency. ... Management and treatment Plummer-Vinson syndrome can be treated effectively with iron supplementation and mechanical dilation.
Plummer–Vinson syndrome Other names Paterson–Brown–Kelly syndrome , Sideropenic dysphagia , Angular stomatitis Specialty Gastroenterology Plummer–Vinson syndrome is a rare disease characterized by difficulty swallowing , iron-deficiency anemia , glossitis , cheilosis and esophageal webs . [1] Treatment with iron supplementation and mechanical widening of the esophagus generally provides an excellent outcome. While exact data about the epidemiology is unknown, this syndrome has become extremely rare. The reduction in the prevalence of Plummer–Vinson syndrome has been hypothesized to be the result of improvements in nutritional status and availability in countries where the syndrome was previously described. [1] It generally occurs in perimenopausal women. ... "Plummer-Vinson syndrome" . Orphanet Journal of Rare Diseases . 1 : 36. doi : 10.1186/1750-1172-1-36 . ... PMID 16978405 . ^ "Plummer-Vinson syndrome: MedlinePlus Medical Encyclopedia" . 2011. ^ a b c Goel, A; Bakshi, SS; Soni, N; Chhavi, N (2017). ... PMID 18031398 . ^ "Plummer-Vinson Syndrome" . PubMed Health . Retrieved 31 January 2012 . ^ synd/1777 at Who Named It?
Blau syndrome is an inflammatory disorder that primarily affects the skin, joints, and eyes. ... Some individuals with Blau syndrome develop kidney disease (nephritis) due to inflammation. ... Causes Blau syndrome results from mutations in the NOD2 gene. ... However, it is unclear how overactivation of the NOD2 protein causes the specific pattern of inflammation affecting the joints, eyes, and skin that is characteristic of Blau syndrome. Learn more about the gene associated with Blau syndrome NOD2 Inheritance Pattern Blau syndrome is inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder. ... In some cases, people with the characteristic features of Blau syndrome do not have a family history of the condition.
Description Blau syndrome is characterized by the triad of granulomatous arthritis, uveitis, and dermatitis. ... Jabs et al. (1985) reported a family in which 4 individuals had a syndrome of granulomatous synovitis and nongranulomatous uveitis. ... Inheritance The transmission pattern of Blau syndrome in the family reported by Blau (1985) was consistent with autosomal dominant inheritance. ... In a 27-year-old Japanese man with systemic granulomatous disease, in whom lack of a family history of the disease led to a diagnosis of 'early-onset sarcoidosis' rather than Blau syndrome, Kanazawa et al. (2004) identified 1 of the same CARD15 mutations (R334W; 605056.0006) that had previously been detected in Blau syndrome. ... Kanazawa et al. (2005) concluded that most patients given a diagnosis of early-onset sarcoidosis or Blau syndrome share a common genetic etiology of CARD15 mutations that cause constitutive NFKB activation, and noted that this supported the long-standing hypothesis that sporadic cases of EOS and familial cases of Blau syndrome represent different types of the same juvenile systemic granulomatosis syndrome.
Blau syndrome is a rare condition characterized mainly by skin rash, arthritis and uveitis . ... It is caused by mutations in the NOD2 gene and is inherited in an autosomal dominant manner. Blau syndrome and early-onset sarcoidosis have the same symptoms and genetic cause, but early-onset sarcoidosis is caused by de novo (new) mutations and occurs sporadically (in individuals with no history of the disorder in the family).
Blau syndrome Other names Arthrocutaneouveal granulomatosis [1] Coarse facial features in a boy with Blau syndrome Specialty Dermatology Multiple, reddish-brown papules coalescing over the right arm in a boy with Blau syndrome Blau Syndrome is an autosomal dominant genetic inflammatory disorder which affects the skin, eyes, and joints. ... "CARD15 mutations in familial granulomatosis syndromes: a study of the original Blau syndrome kindred and other families with large-vessel arteritis and cranial neuropathy". ... Further reading [ edit ] Wouters CH, Maes A, Foley KP et al. Blau syndrome, the prototypic auto-inflammatory granulomatous disease. ... Miceli-Richard C, Lesage S, Rybojad M et al. : CARD!% mutations in Blau syndrome. Nature Genetics 2005; 29: 1920. Wang X, Kuivaniemi H, Bonvita G et al.: CARD 15 Mutations in Familial Granulomatous Syndromes. Arthritis Rheum 2002; 46:3041-3045.
7q11.23 duplication syndrome is a condition that can cause a variety of neurological and behavioral problems as well as other abnormalities. People with 7q11.23 duplication syndrome typically have delayed development of speech and motor skills such as crawling and walking. ... About one-fifth of people with 7q11.23 duplication syndrome experience seizures. Intellectual development varies widely in 7q11.23 duplication syndrome. ... This region is called the Williams-Beuren syndrome critical region (WBSCR) because its deletion causes a different disorder called Williams syndrome, also known as Williams-Beuren syndrome. ... Researchers are studying additional genes in the duplicated region, but none have been definitely linked to any of the specific signs or symptoms of 7q11.23 duplication syndrome. Learn more about the genes and chromosome associated with 7q11.23 duplication syndrome ELN GTF2I chromosome 7 Inheritance Pattern 7q11.23 duplication syndrome is considered to be an autosomal dominant condition, which means one copy of chromosome 7 with the duplication in each cell is sufficient to cause the disorder.
Van der Aa et al. (2009) reported 14 patients with the chromosome 7q11.23 duplication syndrome, including 9 familial and 5 de novo cases. ... Growth hormone deficiency, which had not previously been identified in patients with this syndrome, was identified in 5 probands. ... In addition, the duplication at 7q11.2 of the Williams syndrome region was also seen as a recurrent CNV. ... Pathogenesis Strong et al. (2015) assessed genomewide DNA methylation in whole blood in 20 children with Williams syndrome (194050), 10 children with Dup7 (WBS duplication syndrome), and 15 typically developing children using the Infinium HumanMethylation450 BeadChip array. The authors found that regions that were differentially methylated in both Williams syndrome and Dup7 displayed a significant and symmetrical gene dose-dependent effect, such that Williams syndrome typically showed increased and Dup7 showed decreased DNA methylation.
7q11.23 duplication syndrome is a chromosome abnormality characterized by a variety of neurological and behavioral differences. ... Symptoms may include: motor, speech and language delay, behavior problems, intellectual disability, low muscle tone (hypotonia), an increased head circumference ( macrocephaly ), facial dysmorphism, seizures, brain abnormalities, and heart defects such as enlargement of the blood vessel that carries blood from the heart to the rest of the body (aortic dilatation). 7q11.23 duplication syndrome is caused by a duplication of genetic material on the long (q) arm of chromosome 7. Some cases of 7q11.23 duplication syndrome are inherited in an autosomal dominant manner; however, the majority of affected individuals have a new ( de novo ) duplication.
7q11.23 duplication syndrome Other names dup7 7q11.23 duplication syndrome (also called dup7 or Duplication of the Williams-Beuren Syndrome Critical Region) is a rare genetic syndrome caused by micro-duplication of 1.5-1.8 mega base in section q11.23 of chromosome 7 . This syndrome is characterized by a wide spectrum of neurological, behavior and other medical problems which may appear in different levels of severity. ... (December 2015). "7q11.23 Duplication Syndrome: Physical Characteristics and Natural History" . ... "Aortopathy in the 7q11. 23 microduplication syndrome" (PDF) . American Journal of Medical Genetics Part A . 167 (2). ^ Velleman, Shelley L.; Mervis, Carolyn B. (2011-10-01). "Children with 7q11.23 Duplication Syndrome: Speech, Language, Cognitive, and Behavioral Characteristics and their Implications for Intervention" .
7q11.23 microduplication syndrome is a rare chromosomal anomaly syndrome resulting from the partial duplication of the long arm of chromosome 7 characterized by a highly variable phenotype that typically manifests with mild-moderate intellectual delay (patients could be in the normal range), speech disorders (particularly of expressive language), and distinctive craniofacial features (brachycephaly, broad forehead, straight eyebows, broad nasal tip, short philtrum, thin upper lip and facial asymmetry).
Potocki–Shaffer syndrome Other names Proximal 11p deletion syndrome, DEFECT11 syndrome Potocki–Shaffer syndrome (PSS) , also known as DEFECT11 syndrome or chromosome 11p11.2 deletion syndrome , [1] is a rare contiguous gene syndrome that results from the microdeletion of section 11.2 on the short arm of chromosome 11 (11p11.2). The syndrome has its name from Dr. Lorraine (Lori) Potocki and Dr. ... "Interstitial deletion of 11(p11.2p12): a newly described contiguous gene deletion syndrome involving the gene for hereditary multiple exostoses (EXT2)". ... PMID 8882796 . ^ "Potocki–Shaffer syndrome" . OrphaNet . Paris, France : INSERM . ... Retrieved 26 August 2009 . ^ a b Reference, Genetics Home. "Potocki-Shaffer syndrome" . Genetics Home Reference . Retrieved 2016-09-21 .
Potocki-Shaffer syndrome is a disorder that affects development of the bones, nerve cells in the brain, and other tissues. ... Frequency Potocki-Shaffer syndrome is a rare condition, although its prevalence is unknown. Fewer than 100 cases have been reported in the scientific literature. Causes Potocki-Shaffer syndrome (also known as proximal 11p deletion syndrome) is caused by a deletion of genetic material from the short (p) arm of chromosome 11 at a position designated 11p11.2. ... The loss of multiple genes within the deleted region causes the varied signs and symptoms of Potocki-Shaffer syndrome. In particular, deletion of the EXT2 , ALX4 , and PHF21A genes are associated with several of the characteristic features of Potocki-Shaffer syndrome. ... The loss of additional genes in the deleted region likely contributes to the other features of Potocki-Shaffer syndrome. Learn more about the genes and chromosome associated with Potocki-Shaffer syndrome ALX4 EXT2 PHF21A chromosome 11 Inheritance Pattern Potocki-Shaffer syndrome follows an autosomal dominant inheritance pattern, which means a deletion of genetic material from one copy of chromosome 11 is sufficient to cause the disorder.
A number sign (#) is used with this entry Potocki-Shaffer syndrome is a contiguous gene deletion syndrome involving genes on chromosome 11p11.2. Description Potocki-Shaffer syndrome is a rare contiguous gene deletion syndrome due to haploinsufficiency of the 11p12-p11.2 region and is characterized by craniofacial abnormalities, developmental delay, intellectual disability, multiple exostoses (168500), and biparietal foramina (605957) (summary by Swarr et al., 2010). Clinical Features Bartsch et al. (1996) described a contiguous gene syndrome due to deletion in the proximal short arm of chromosome 11 in 8 patients belonging to 4 families. ... McGaughran et al. (1995) reported a patient with the combination of 2 deletion syndromes, WAGR (194072) and Potocki-Shaffer syndrome. ... Wakui et al. (2005) constructed a panel of 11p deletions using cell lines derived from 10 patients with Potocki-Shaffer syndrome, including 6 patients who were newly identified.
Potocki-Shaffer syndrome is a contiguous gene deletion syndrome associated with deletions in a specific region of chromosome 11 (11p11.2). The characteristic features of Potocki-Shaffer syndrome include openings in the two bones that form the top and sides of the skull (enlarged parietal foramina), multiple benign (non-cancerous) bone tumors called exostoses, intellectual disability, developmental delay, a distinctive facial appearance, autism spectrum disorder , and problems with vision and hearing. In some cases, individuals with the syndrome may have a defect in the heart, kidneys, or urinary tract. ... The features of Potocki-Shaffer syndrome result from the loss of several genes on the short (p) arm of chromosome 11 . ... Individuals with such a deletion have signs and symptoms of both Potocki-Shaffer syndrome and WAGR syndrome . [ A referral to an early childhood intervention and developmental-behavioral specialist and evaluation for vision and hearing problems at the time of diagnosis is recommended.
A rare partial autosomal monosomy characterized by global developmental delay, intellectual disability, multiple cartilaginous exostoses, and craniofacial anomalies (such as brachycephaly, biparietal foramina, large fontanels, craniosynostosis, ptosis, epicanthic folds, prominent nasal bridge with broad, depressed nasal tip, hypoplastic nares, short philtrum, downturned upper lip, and micrognathia). Additional reported features include behavioral abnormalities, myopia, strabismus, and sensorineural hearing loss, among others.
Partington syndrome usually occurs in males; when it occurs in females, the signs and symptoms are often less severe. The intellectual disability associated with Partington syndrome usually ranges from mild to moderate. ... Recurrent seizures (epilepsy) may also occur in Partington syndrome. Focal dystonia of the hands is a feature that distinguishes Partington syndrome from other intellectual disability syndromes. ... Causes Partington syndrome is caused by mutations in the ARX gene. ... The mosaic nature of these genetic changes lead to relatively mild features of Partington syndrome.
X-linked reticulate pigmentary disorder is an extremely rare skin disease described in only four families to date and characterized in males by diffuse reticulate brown hyperpigmentated skin lesions developing in early childhood and a variety of systemic manifestations (recurrent pneumonia, corneal opacification, gastrointestinal inflammation, urethral stricture, failure to thrive, hypohidrosis, digital clubbing, and unruly hair and flared eyebrows), while in females, there is only cutaneous involvement with the development in early childhood of localized brown hyperpigmented skin lesions following the lines of Blaschko. This disease was first considered as a cutaneous amyloidosis, but amyloid deposits are an inconstant feature.
Description Partington syndrome is an X-linked developmental disorder characterized by mental retardation and variable movement disturbances. Partington syndrome is part of a phenotypic spectrum of disorders caused by mutation in the ARX gene comprising a nearly continuous series of developmental disorders ranging from hydranencephaly and lissencephaly (LISX2; 300215) to Proud syndrome (300004) to infantile spasms without brain malformations (EIEE1; 308350) to nonsyndromic mental retardation (300419). ... Frints et al. (2002) described 2 Belgian brothers with Partington syndrome. The brothers had a delayed and aberrant grasp reflex which resulted in feeding difficulties in early childhood. ... Szczaluba et al. (2006) reported 18 individuals from 5 families with an X-linked mental retardation syndrome due to a hemizygous 24-bp duplication in the ARX gene. The phenotype was variable but most consistent with Partington syndrome. All patients had intellectual impairment.
Partington syndrome is a form of syndromic X-linked mental retardation (S-XLMR) characterised by the association of mild to moderate intellectual deficit, dysarthria and dystonic hand movements. So far, less than 20 cases have been described in the literature. The syndrome is caused by mutations in the Aristaless-related homeobox ( ARX ) gene (Xp22.13).
Partington syndrome is a rare neurological condition that is primarily characterized by mild to moderate intellectual disability and dystonia of the hands. Other signs and symptoms may include dysarthria, behavioral abnormalities, recurrent seizures and/or an unusual gait (style of walking). Partington syndrome usually occurs in males; when it occurs in females, the signs and symptoms are often less severe.
Nager syndrome is a rare condition that mainly affects the development of the face, hands, and arms. ... Individuals with Nager syndrome have bone abnormalities in their hands and arms. ... Causes More than half of cases of Nager syndrome are caused by mutations in the SF3B4 gene. ... SF3B4 gene mutations that cause Nager syndrome prevent the production of functional SAP49 protein. ... Learn more about the gene associated with Nager syndrome SF3B4 Inheritance Pattern When caused by mutations in the SF3B4 gene, Nager syndrome follows an autosomal dominant inheritance pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
A congenital malformation syndrome characterized by mandibulofacial dystosis (malar hypoplasia, micrognathia, external ear malformations) and variable preaxial limb defects. ... Otologic and oral anomalies frequently lead to bilateral conductive hearing loss, speech difficulties and upper respiratory airways obstruction. Most Nager syndrome individuals have normal vision and intelligence. ... Differential diagnosis Differential diagnosis may include mandibulofacial dysostosis syndromes such as Treacher-Collins syndrome, and other acrofacial dysostoses (AFD) such as the AFD Catania type, the AFD Palagonia type, the AFD Genee-Wiedemann type, the AFD Rodriquez type as well as mandibulofacial dysostosis with microcephaly (see these terms). ... Antenatal diagnosis Antenatal diagnosis can be performed by ultrasonography or molecular testing of SF3B4 . Genetic counseling Nager syndrome is likely genetically heterogenous with confirmed autosomal dominant inheritance, but autosomal recessive inheritance is suspected based on sibling recurrence in consanguineous families.
The presence of anterior upper-limb defects and the typical lack of lower-limb involvement distinguishes Nager syndrome from Miller syndrome (263750), another rare AFD; however, distinguishing Nager syndrome from other AFDs, including Miller syndrome, can be challenging (summary by Bernier et al., 2012). ... The disorder reported by Walker (1974) in sibs whose parents were normal may have been Nager syndrome. Weinbaum et al. (1981) described a kindred in which the proband had classic Nager syndrome and 5 other persons covering 4 generations showed lesser expression. ... The differentiation from AFD with postaxial defects (263750), the hemifacial microsomia/Goldenhar radial defect syndrome, and other syndromes was discussed. ... See 263750 for the Miller acrofacial syndrome in which postaxial limb changes (rather than preaxial as in Nager syndrome) are associated with facial dysostosis. ... Scapoli et al. (2003) described an infant with Nager syndrome and a chromatid gap within band 3p14.
Nager acrofacial dysostosis Other names Nager syndrome, mandibulofacial dysostosis with preaxial limb anomalies [1] Nager acrofacial dysostosis is inherited in an autosomal dominant manner. [2] Nager acrofacial dysostosis , also known as Nager syndrome , is a genetic disorder which displays several or all of the following characteristics: underdevelopment of the cheek and jaw area , down-sloping of the opening of the eyes , lack or absence of the lower eyelashes , kidney or stomach reflux , hammer toes , shortened soft palate , lack of development of the internal and external ear , possible cleft palate , underdevelopment or absence of the thumb , hearing loss (see hearing loss with craniofacial syndromes ) and shortened forearms , as well as poor movement in the elbow , and may be characterized by accessory tragi . [3] Occasionally, affected individuals develop vertebral anomalies such as scoliosis . ... Most cases tend to be sporadic. Nager syndrome shares many characteristics with five other craniofacial syndromes: Miller , Treacher Collins , Pierre Robin , Genee–Wiedemann and Franceschetti–Zwahlen–Klein . Contents 1 Genetics 2 Treatment 3 See also 4 References 5 External links Genetics [ edit ] Nager syndrome is thought to be caused by haploinsufficiency of the spliceosomal factor SF3B4 . [4] Treatment [ edit ] This section is empty. You can help by adding to it . ( August 2017 ) See also [ edit ] Dermoid cyst References [ edit ] ^ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Nager syndrome" . www.orpha.net . Retrieved 27 April 2019 . ^ "OMIM Entry - # 154400 - ACROFACIAL DYSOSTOSIS 1, NAGER TYPE; AFD1" . omim.org . ... "Haploinsufficiency of SF3B4, a Component of the Pre-mRNA Spliceosomal Complex, Causes Nager Syndrome" . American Journal of Human Genetics . 90 (5): 925–33. doi : 10.1016/j.ajhg.2012.04.004 .
Nager acrofacial dysostosis is a genetic disorder that affects the limbs and face. The signs and symptoms of Nager acrofacial dysostosis vary among affected individuals, even among those in the same family. Treatment is tailored to the individual based upon their specific needs. This condition is caused by mutations in the SF3B4 gene. While most cases are sporadic (occurring in families with no prior history of the disorder), autosomal dominant and autosomal recessive inheritance have been reported.
Hennekam syndrome Other names ennekam lymphangiectasia–lymphedema syndrome, intestinal lymphagiectasia–lymphedema–mental retardation syndrome [1] Hennekam syndrome is inherited in an autosomal dominant manner Hennekam syndrome also known as intestinal lymphagiectasia–lymphedema–mental retardation syndrome , [1] is an autosomal recessive disorder consisting of intestinal lymphangiectasia, facial anomalies, peripheral lymphedema , and mild to moderate levels of growth and intellectual disability . [1] [2] It is also known as "lymphedema-lymphangiectasia-mental retardation syndrome". [3] Hennekam Syndrome is subdivided according to the causative genetic lesion, most (or all) of which are affecting the VEGF-C/VEGFR-3 signaling pathway: Type 1 (mutations in CCBE1 ) [4] Type 2 (mutations in FAT4 ) [5] Type 3 (mutations in ADAMTS3 ) [6] The first recognition of a genetic association was with CCBE1 , published by its namesake, Raoul Hennekam. [7] The molecular mechanism of the lymphedema phenotype in CCBE1-associated cases was identified as a diminished ability of the mutated CCBE1 to accelerate and focus the activation of the primary lymphangiogenic growth factor VEGF-C . [8] Mutations in the FAT4 gene had previously been only associated with van Maldergem syndrome, but the pathogenetic molecular mechanism and the function of FAT4 within lymphangiogenesis are still unknown. ... Hennekam lymphangiectasia–lymphedema syndrome. In: Catalogue for Transmission Genetics in Arabs [PDF]. Centre for Arab Genomic Studies; 2005. ^ Erkan T, Kutlu T, Çullu F et al. [Hennekam syndrome]. Arch Pediatr . 1998;5(12):1344–6. ... "Lymphedema-lymphangiectasia-mental retardation (Hennekam) syndrome: a review". Am. J. Med. Genet . 112 (4): 412–21. doi : 10.1002/ajmg.10707 . ... PMID 19935664 . ^ Alders M, Al-Gazali L, Cordeiro I, et al. (June 2014). "Hennekam syndrome can be caused by FAT4 mutations and be allelic to Van Maldergem syndrome".
A number sign (#) is used with this entry because of evidence that Hennekam lymphangiectasia-lymphedema syndrome-3 (HKLLS3) is caused by homozygous or compound heterozygous mutation in the ADAMTS3 gene (605011) on chromosome 4q13. Description Hennekam lymphangiectasia-lymphedema syndrome-3 (HKKLLS3) is characterized by widespread congenital edema that is more severe in more dependent areas of the body. ... For a discussion of genetic heterogeneity of Hennekam lymphangiectasia-lymphedema syndrome, see HKLLS1 (235510). Clinical Features Brouillard et al. (2017) reported a sister and brother of western European origin born with congenital lymphedema of the lower extremities. ... In a male infant with Hennekam lymphangiectasia-lymphedema syndrome-3, Scheuerle et al. (2018) identified a homozygous nonsense mutation in the ADAMTS3 gene (R94X; 605011.0003).
A rare syndromic lymphedema characterized by the association of primary lymphedema, intestinal lymphangiectasia, intellectual deficit and unusual facial characteristics. ... Other features include mild growth retardation, delayed puberty, tooth anomalies, gingival hypertrophy, seizures, and blood vessel anomalies. Etiology The syndrome can be caused by bi-allelic variants in CCBE1 (18q21.32) or FAT4 (4q28.1) or ADAMTS3 (4q13.3) can cause Hennekam syndrome. ... Differential diagnosis The differential diagnosis includes other entities associated with congenital lymphedema such as Noonan syndrome, Milroy syndrome and cholestasis-lymphedema syndrome, and Van Maldergem syndrome. Antenatal diagnosis Prenatal genetic testing is possible in families with known mutations. Genetic counseling The syndrome is transmitted as an autosomal recessive trait and genetic counseling is recommended.
He had a face typical of Hennekam syndrome, including flat nasal bridge, hypertelorism, small mouth and tooth anomalies, but did not have mental retardation or severe lymphedema. Yasunaga et al. (1993) suggested that the child had a mild form of Hennekam syndrome. Study of the family in 3 generations suggested that heterozygotes may have some of the facial features. ... Forzano et al. (2002) proposed that the patient had a severe form of Hennekam syndrome. Van Balkom et al. (2002) reported 8 patients with Hennekam syndrome and compared their findings with those of the 16 previously reported cases. ... Mapping In a large consanguineous Dutch pedigree with Hennekam syndrome, Alders et al. (2009) performed homozygosity mapping and identified a 5.7-Mb homozygous region on chromosome 18q21; additional homozygosity mapping in 2 unrelated probands with Hennekam syndrome from consanguineous families revealed several stretches of homozygosity, including an overlapping 0.5-Mb interval on 18q21 containing 4 genes. ... Alders et al. (2009) noted that mutations were detected in only 5 (23%) of 22 families with Hennekam syndrome, indicating genetic heterogeneity.
Biallelic mutation in the FAT4 gene can also cause Van Maldergem syndrome-2 (VMLDS2; 615546), a distinct disorder that shows overlapping features with HKLLS. ... For a discussion of genetic heterogeneity of Hennekam lymphangiectasia-lymphedema syndrome, see HKLLS1 (235510). Clinical Features Hennekam et al. (1989) described a syndrome of intestinal lymphangiectasia with severe lymphedema of the limbs, genitalia, and face, and severe mental retardation. ... The facial appearance was Oriental. Down syndrome had been suspected in some of the patients. ... Hennekam et al. (1989) reviewed genetic syndromes with lymphangiectasia and lymphedema as features. ... Subsequent mutations were identified in 4 of 24 Hennekam syndrome patients who underwent direct sequencing of the FAT4 gene.
Hennekam syndrome is a rare condition that affects the lymphatic system. ... Other common features include intellectual disability, growth delay, respiratory problems, camptodactyly (permanently bent fingers and toes) and cutaneous syndactyly (fusion of the skin between the fingers and toes). Hennekam syndrome is caused by changes (mutations) in the CCBE1 or FAT4 genes and is inherited in an autosomal recessive manner.
Ramsay Hunt syndrome Other names Facial nerve palsy due to herpes zoster infection Specialty Neurology Three different neurological syndromes carry the name of Ramsay Hunt syndrome . Their only connection is that they were all first described by the famous neurologist James Ramsay Hunt (1872–1937). Ramsay Hunt syndrome type 1 , also called Ramsay Hunt cerebellar syndrome , is a rare form of cerebellar degeneration which involves myoclonic epilepsy, progressive ataxia , tremor , and a dementing process. [1] Ramsay Hunt syndrome type 2 is the reactivation of herpes zoster in the geniculate ganglion . ... "On herpetic inflammations of the geniculate ganglion: a new syndrome and its complications" . Journal of Nervous and Mental Disease . 34 (2): 73–96. doi : 10.1097/00005053-190702000-00001 . ^ Sweeney, C.J.; Gilden, D.H. (August 2001). "Ramsay Hunt Syndrome" . Journal of Neurology, Neurosurgery, and Psychiatry . 71 (2): 149–54. doi : 10.1136/jnnp.71.2.149 . ... PMID 11459884 . ^ Pearce, J.M.S. (2007). "Some Syndromes of James Ramsay Hunt" . Practical Neurology . 7 (3): 182–185.
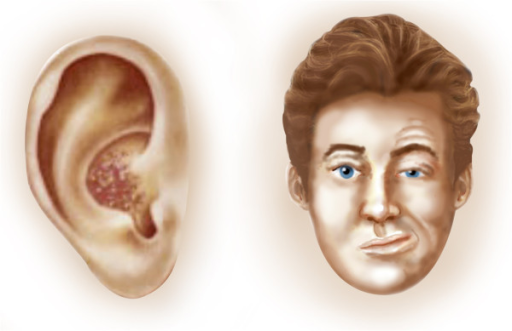
Overview Ramsay Hunt syndrome (herpes zoster oticus) occurs when a shingles outbreak affects the facial nerve near one of your ears. ... Ramsay Hunt syndrome is caused by the same virus that causes chickenpox. ... It's more common in older adults, typically affecting people older than 60. Ramsay Hunt syndrome is rare in children. Ramsay Hunt syndrome isn't contagious. ... The facial weakness caused by Ramsay Hunt syndrome may make it difficult for you to close your eyelid. ... The pain associated with Ramsay Hunt syndrome can be severe. Prescription pain medications may be needed.
For other uses, see Ramsay Hunt syndrome . Ramsay Hunt syndrome (RHS) type 2 Other names Herpes zoster oticus Specialty Infectious disease Ramsay Hunt syndrome type 2 , also known as herpes zoster oticus , is a disorder that is caused by the reactivation of varicella zoster virus in the geniculate ganglion , a nerve cell bundle of the facial nerve . [1] Ramsay Hunt syndrome type 2 typically presents with inability to move many facial muscles, pain in the ear, taste loss on the front of the tongue, dry eyes and mouth, and a vesicular rash. ... Involvement of the trigeminal nerve can cause numbness of the face. [ citation needed ] Pathophysiology [ edit ] Ramsay Hunt syndrome type 2 refers to shingles of the geniculate ganglion. ... (August 2001). "Ramsay Hunt syndrome" . Journal of Neurology, Neurosurgery, and Psychiatry . 71 (2): 149–54. doi : 10.1136/jnnp.71.2.149 . ... "Correlation between MRI and Operative Findings in Bell's Palsy and Ramsay Hunt Syndrome" . Yonsei Medical Journal . 48 (6): 963–968. doi : 10.3349/ymj.2007.48.6.963 . ... PMID 20824789 . ^ NINDS Herpes Zoster Oticus Information Page at NINDS ^ synd/2246 at Who Named It? ^ "The Ramsay Hunt syndrome" . Proceedings of the Royal Society of Medicine . 47 (5): 371–384.
A rare infectious disease characterized by herpes zoster oticus associated with peripheral facial nerve palsy, often also with other cranial nerve lesions. Patients present with a painful erythematous vesicular rash in and around one ear and facial paralysis on the same side. Other frequent manifestations include hearing loss, tinnitus, vertigo, nausea, vomiting, and nystagmus.
Herpes zoster oticus is a common complication of shingles , an infection caused by the varicella-zoster virus (which is the virus that also causes chickenpox ). Shingles occurs in people who have had chickenpox and in whom the varicella-zoster virus becomes active again. Herpes zoster oticus is caused by the spread of the virus to facial nerves and can cause intense ear pain; a rash around the ear, mouth, face, neck, and scalp; and paralysis of the face. Other symptoms may include hearing loss, vertigo (feeling that the room is spinning), tinnitus (hearing abnormal sounds), nausea, vomiting, loss of taste in the tongue, and dry mouth and eyes. Some cases of herpes zoster oticus do not require treatment, but when treatment is needed, pain medications, antiviral drugs or corticosteroids may be prescribed.
Specifically, low-set ears are defined as outer ears positioned two or more standard deviations lower than the population average. [1] Low-set ears can be associated with conditions such as: Down syndrome [2] Turner syndrome Noonan syndrome [3] Patau syndrome [4] DiGeorge syndrome [5] Cri du chat syndrome Edwards syndrome Fragile X syndrome Okamoto syndrome It is usually bilateral, but it can be unilateral in Goldenhar syndrome . [6] See also [ edit ] LEOPARD syndrome References [ edit ] ^ Sivan Y, Merlob P, Reisner SH (June 1983). ... PMC 1049049 . PMID 6876114 . ^ "Down's Syndrome" . ^ Sanchez-Cascos, A. (1983). "The Noonan syndrome". European Heart Journal . 4 (4): 223–229. doi : 10.1093/oxfordjournals.eurheartj.a061452 .
Gleich's syndrome Other names Episodic angioedema with eosinophilia Gleich's syndrome is a rare disease in which the body swells up episodically ( angioedema ), associated with raised antibodies of the IgM type and increased numbers of eosinophil granulocytes , a type of white blood cells , in the blood ( eosinophilia ). It was first described in 1984. [1] Its cause is unknown, but it is unrelated to capillary leak syndrome (which may cause similar swelling episodes) and eosinophilia-myalgia syndrome (which features eosinophilia but alternative symptoms). Some studies have shown that edema attacks are associated with degranulation (release of enzymes and mediators from eosinophils), and others have demonstrated antibodies against endothelium (cells lining blood vessels) in the condition. [2] Gleich's syndrome is not a form of the idiopathic hypereosinophilic syndrome in that there is little or no evidence that it leads to organ damage. ... It is suggested that most forms of Gleich's syndrome are due to a similar aberrant T cell mechanism and are a subtype of lymphocyte-variant eosinophilia. [3] Gleich syndrome has a good prognosis. ... PMID 6727934 . ^ a b Emonet S, Kaya G, Hauser C (2000). "Gleich's syndrome" . Ann Dermatol Venereol . 127 (6–7): 616–8.
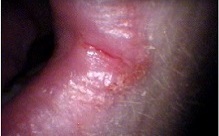
Hailey-Hailey disease is a genetic skin disease that causes blistering. Signs and symptoms include a painful rash and blistering in skin folds, such as the armpits, groin, neck, under the breasts, and between the buttocks. Secondary bacterial infections are not uncommon. Symptoms are often worse in summer months due to heat, sweating, and friction. Hailey-Hailey disease is caused by mutations in the ATP2C1 gene and is inherited in an autosomal dominant manner. Treatment focuses on reducing symptoms and preventing flares, and may include topical medication, laser, and other procedures.
Hailey-Hailey disease, also known as benign chronic pemphigus, is a rare skin condition that usually appears in early adulthood. The disorder is characterized by red, raw, and blistered areas of skin that occur most often in skin folds, such as the groin, armpits, neck, and under the breasts. These inflamed areas can become crusty or scaly and may itch and burn. The skin problems tend to worsen with exposure to moisture (such as sweat), friction, and hot weather. The severity of Hailey-Hailey disease varies from relatively mild episodes of skin irritation to widespread, persistent areas of raw and blistered skin that interfere with daily activities.
A number sign (#) is used with this entry because of evidence that Hailey-Hailey disease is caused by heterozygous mutation in the ATP2C1 gene (604384) on chromosome 3q22. Description Hailey-Hailey disease, also known as benign chronic pemphigus, is a rare autosomal dominant cutaneous disorder that usually becomes manifest in the third or fourth decade of life with erythema, vesicles, and erosions involving the body folds, particularly the groin and axillary regions. Other sites of the body, such as the neck, perianal, and submammary regions, may likewise be affected (summary by Poblete-Gutierrez et al., 2004). This disorder was first described by the dermatologist brothers Hailey and Hailey (1939). Clinical Features Loewenthal (1959) thought that pyogenic bacteria act as a precipitating factor.
Benign chronic familial pemphigus of Hailey-Hailey is characterized by rhagades mostly located in the armpits, inguinal and perineal folds (scrotum, vulva). Epidemiology Prevalence is unknown. Clinical description Skin lesions appear during adolescence or more often at the age of 30-40 years; they are relapsing and recurrent. Lesions can be complicated by heat, rubbing or superinfections. Etiology Mutations in the ATP2C1 gene (localised to 3q21-q24), encoding a calcium pump, cause the disease by impairing epidermal keratinocyte adhesion. Diagnostic methods Histopathological analysis of the lesions shows suprabasal acantholysis of epidermal cells. Genetic counseling Benign chronic familial pemphigus is transmitted as a dominant trait, with incomplete penetrance.