Cain and Fletcher (2010) report a case of metal fume fever that was diagnosed only by taking a full occupational history and by close collaboration between primary and secondary health care personnel. [12] Physical symptoms vary among persons exposed, depending largely upon the stage in the course of the syndrome during which examination occurs. ... Chest X-ray abnormalities may also be present. [10] An interesting feature of metal fume fever involves rapid adaptation to the development of the syndrome following repeated metal oxide exposure. ... It does not prevent you getting metal fume fever." [14] Prevention [ edit ] Prevention of metal fume fever in workers who are at risk (such as welders ) involves avoidance of direct contact with potentially toxic fumes, improved engineering controls (exhaust ventilation systems), personal protective equipment (respirators), and education of workers regarding the features of the syndrome itself and proactive measures to prevent its development. [ citation needed ] In some cases, the product's design may be changed so as to eliminate the use of risky metals. ... PMID 10931787 . v t e Occupational safety and health Occupational diseases and injuries Acrodynia Asbestosis Asthma Barotrauma Berylliosis Brucellosis Byssinosis ("brown lung") Chalicosis Chimney sweeps' carcinoma Chronic solvent-induced encephalopathy Coalworker's pneumoconiosis ("black lung") Concussions in sport Decompression sickness De Quervain syndrome Erethism Exposure to human nail dust Farmer's lung Fiddler's neck Flock worker's lung Glassblower's cataract Golfer's elbow Hearing loss Hospital-acquired infection Indium lung Laboratory animal allergy Lead poisoning Mesothelioma Metal fume fever Mule spinners' cancer Noise-induced hearing loss Phossy jaw Pneumoconiosis Radium jaw Repetitive strain injury Silicosis Silo-filler's disease Sports injury Surfer's ear Tennis elbow Tinnitus Writer's cramp Occupational hygiene Occupational hazard Biological hazard Chemical hazard Physical hazard Psychosocial hazard Hierarchy of hazard controls Prevention through design Exposure assessment Occupational exposure limit Occupational epidemiology Workplace health surveillance Professions Environmental health Industrial engineering Occupational health nursing Occupational health psychology Occupational medicine Occupational therapist Safety engineering Agencies and organizations Canadian Centre for Occupational Health and Safety European Agency for Safety and Health at Work UK Health and Safety Executive International Labour Organization US National Institute for Occupational Safety and Health US Occupational Safety and Health Administration National Institute for Safety and Health at Work (Spain) World Health Organization Standards Bangladesh Accord ISO 45001 Occupational Safety and Health Convention, 1981 Worker Protection Standard (US) Working Environment Convention, 1977 Safety Checklist Code of practice Contingency plan Diving safety Emergency procedure Emergency evacuation Hazard Hierarchy of hazard controls Hazard elimination Administrative controls Engineering controls Hazard substitution Personal protective equipment Job safety analysis Lockout-tagout Permit To Work Operations manual Redundancy (engineering) Risk assessment Safety culture Standard operating procedure Legislation Diving regulations Occupational Safety and Health Act (United States) See also Environment, health and safety Environmental toxicology Ergonomics Health physics Indoor air quality International Chemical Safety Card National Day of Mourning (Canadian observance) Process safety management Public health Risk management Safety data sheet Toxic tort Workers' compensation Category Occupational diseases Journals Organizations Commons Glossary
Specialty Nephrology Prognosis 1- and 5-year survival rates are estimated to be 45% and 35%, respectively Calciphylaxis , also known as calcific uremic arteriolopathy ( CUA ) or “Grey Scale”, is a rare painful syndrome of calcification of the small blood vessels located within the fatty tissue and deeper layers of the skin , blood clots , and the death of skin cells due to too little blood flow . [1] It is seen mostly in people with end-stage kidney disease but can occur in the earlier stages of chronic kidney disease and rarely in people with normally functioning kidneys . [1] It results in chronic non-healing wounds and is usually fatal. ... Similar extraskeletal calcifications are observed in some people with high levels of calcium in the blood , including people with milk-alkali syndrome , sarcoidosis , primary hyperparathyroidism , and hypervitaminosis D . ... External links [ edit ] Classification D ICD - 9-CM : 275.49 MeSH : D002115 DiseasesDB : 1897 SNOMED CT : 237900002 External resources eMedicine : derm/555 Orphanet : 280062 v t e Electrolyte imbalances Sodium High Salt poisoning Low Hypotonic Isotonic Cerebral salt-wasting syndrome Potassium High Low Chloride High Low Calcium High Low Symptoms and signs Chvostek sign Trousseau sign Milk-alkali syndrome Disorders of calcium metabolism Calcinosis ( Calciphylaxis , Calcinosis cutis ) Calcification ( Metastatic calcification , Dystrophic calcification ) Familial hypocalciuric hypercalcemia Phosphate High Low Magnesium High Low
Overview Calciphylaxis (kal-sih-fuh-LAK-sis) is a serious, uncommon disease in which calcium accumulates in small blood vessels of the fat and skin tissues. Calciphylaxis causes blood clots, painful skin ulcers and may cause serious infections that can lead to death. People who have calciphylaxis usually have kidney failure and are on dialysis or have had a kidney transplant. The condition can also occur in people without kidney disease. Symptoms Signs and symptoms of calciphylaxis include: Large purple net-like patterns on skin Deep, very painful lumps that ulcerate creating open sores with black-brown crust that fails to heal — typically in skin areas with high fat content, such as the stomach and thigh, although they can occur anywhere Infections from wounds that don't heal Causes The exact cause of calciphylaxis is unknown, but recent studies have revealed that most people with the condition have abnormalities in blood-clotting factors. Blood-clotting factors are substances in your blood that help stop bleeding.
An Orphanet summary for this disease is currently under development. However, other data related to the disease are accessible from the Additional Information menu located on the right side of this page.
Calciphylaxis is a disease in which blood vessels (veins and arteries) become blocked by a build-up of calcium in the walls of the vessels, preventing blood from flowing to the skin or internal organs. The lack of blood flow ( ischemia ) damages healthy tissue and causes it to die ( necrosis ). The most obvious and frequent symptom of calciphylaxis is damage to the skin, as ulcers can develop and become infected easily. Calciphylaxis can also affect fat tissue, internal organs, and skeletal muscle , causing infections, pain, and organ failure. These symptoms are often irreversible, and many individuals with calciphylaxis may not survive more than a few months after they are diagnosed due to infection that spreads throughout the body ( sepsis ), or organ failure.
Risk factors These factors increase the risk of osteosarcoma: Previous treatment with radiation therapy Other bone disorders, such as Paget's disease of bone and fibrous dysplasia Certain inherited or genetic conditions, including hereditary retinoblastoma, Bloom syndrome, Li-Fraumeni syndrome, Rothmund-Thomson syndrome and Werner syndrome Complications Complications of osteosarcoma and its treatment include: Cancer that spreads (metastasizes).
Osteosarcoma is a feature of Li-Fraumeni syndrome-1 (LFS1; 151623), caused by mutation in the TP53 gene (191170), and of Li-Fraumeni syndrome-2 (LFS2; 609265), caused by mutation in the CHEK2 gene (604373). ... Osteosarcoma is a component of the acronymically designated OSLAM syndrome (165660). Clinical Features Harmon and Morton (1966) reported osteogenic sarcoma in 4 sibs, with onset at 11, 15, 20, and 22 years.
Bone dysplasias, including Paget's disease of bone , fibrous dysplasia , enchondromatosis , and hereditary multiple exostoses , increase the risk of osteosarcoma. Li–Fraumeni syndrome (germline TP53 mutation) is a predisposing factor for osteosarcoma development. Rothmund–Thomson syndrome (i.e. autosomal recessive association of congenital bone defects, hair and skin dysplasias, hypogonadism , and cataracts) is associated with increased risk of this disease.
Osteosarcoma is a primary malignant tumour of the skeleton characterised by the direct formation of immature bone or osteoid tissue by the tumour cells. Epidemiology Classic osteosarcoma is a rare (0.2% of all malignant tumours) highly malignant tumour, with an estimated incidence of 3 cases/million population/year. Clinical description Osteosarcoma arises predominantly in the long bones and rarely in the soft tissues. The age at presentation ranges from 10 to 25 years of age. Diagnostic methods Plain radiographs, computed tomography, magnetic resonance imaging, angiography and dynamic bone scintigraphy are used for diagnosis, evaluation the extent of tumour involvement and for making decisions about the type of operation and, if necessary, the type of reconstruction required. Management and treatment In the past, all patients with osteosarcoma were treated by amputation but the cure rate was under 10% and almost all patients died within a year from diagnosis.
Osteosarcoma is the most common type of bone cancer. The average age at diagnosis is 15. Boys and girls have a similar incidence of this tumor until late adolescence, at which time boys are more commonly affected. In rare cases, osteosarcoma occurs in adults. Although osteosarcoma tends to occur in the larger bones, such as the shin (near the knee), thigh (near the knee) and upper arm (near the shoulder), it can occur in any bone. A number of variants of osteosarcoma exist, including conventional types (osteoblastic, chondroblastic, and fibroblastic), telangiectatic, multifocal, parosteal, and periosteal. The cause of osteosarcoma is not known. In some cases, it runs in families, and at least one gene has been linked to increased risk.
Bartram et al. (1982) observed subacute sclerosing panencephalitis in a brother and sister of nonconsanguineous parents of 11 children living in rural Turkey. An interval of 4 years separated onset of symptoms in the 2 children. Fibroblast interferon had no beneficial effect. Neuro - Subacute sclerosing panencephalitis Inheritance - Autosomal recessive ▲ Close
A chronic progressive encephalitis that develops a few years after measles infection and presents with a demyelination of the cerebral cortex. Epidemiology Due to the measles immunization, nowadays cases of SSPE are very rare. In the United States, less than 10 cases/year are reported. However, in some countries like India, over 20 cases/per million people/per year are reported. Males are more often affected than females (3:1). Clinical description SSPE occurs primarily in children and young adults, approximately 2-8 years after the initial infection. Patients present with a history of measles infection (though the person seems to have fully recovered from the illness).
Subacute sclerosing panencephalitis (SSPE) a rare condition that is caused by a measles infection acquired earlier in life. Signs and symptoms of the condition primarily affect the central nervous system and often develop approximately 7 to 10 years after a person recovers from the measles. Affected people may initially experience behavioral changes, dementia, and disturbances in motor function. In the late stages of the disease, affected people often progress to a comatose state, and then to a persistent vegetative state. Ultimately, many people with SSPE succumb to fever, heart failure, or the brain's inability to continue controlling the autonomic nervous system .
It can also be seen in syringomyelia , Friedreich's ataxia , spina bifida , kyphoscoliotic Ehlers–Danlos syndrome (kEDS), and Duchenne muscular dystrophy due to asymmetric weakening of the paraspinal muscles. ... Sometimes, a traumatic injury can also lead to its development. [ citation needed ] Further, there are many idiopathic occurrences of kyphoscoliosis where the exact cause is not very well known but is suspected to be caused by genetic factors. [ citation needed ] Diagnosis [ edit ] Kyphoscoliosis is one of the main criteria in kyphoscoliotic Ehlers–Danlos syndrome . It is caused by mutation in the PLOD1 gene and/or FKBP14 [1] gene. ... See also [ edit ] Lordosis Pott's disease Ehlers-Danlos Syndrome References [ edit ] ^ https://ghr.nlm.nih.gov/gene/FKBP14 ^ "EDS Types" . ... Retrieved 22 May 2018 . ^ Brady AF, Demirdas S, Fournel-Gigleux S, Ghali N, Giunta C, Kapferer-Seebacher I, Kosho T, Mendoza-Londono R, Pope MF, Rohrbach M, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Zschocke J, Malfait F. 2017. "The Ehlers–Danlos syndromes, rare types". American Journal of Medical Genetics Part C: Seminars in Medical Genetics 175C:70–115.
The authors suggested that generalized gangliosidosis, which they also called 'neurovisceral lipidosis,' may be closely related to the Hurler syndrome, which it resembles clinically and radiologically. Grossman and Danes (1968) demonstrated x-ray features resembling those of Hurler syndrome, increased synthesis and storage of mucopolysaccharides by skin fibroblasts, and marked metachromasia of fibroblasts in both parents. ... Hanson et al. (2003) described 2 infants with extensive dermal melanocytosis in association with GM1-gangliosidosis type I and Hurler syndrome, respectively. Clinically, dermal melanocytosis associated with lysosomal storage disease is characterized by extensive blue cutaneous pigmentation with dorsal and ventral distribution, indistinct borders, and persistent and/or 'progressive' behavior. ... The most common lysosomal storage disease associated with dermal melanocytosis was Hurler syndrome (24 of 39 cases), followed by GM1-gangliosidosis (11 of 39 cases).
A number sign (#) is used with this entry because type III GM1-gangliosidosis is caused by mutation in the gene encoding beta-galactosidase-1 (GLB1; 611458). For a general discussion of classification and phenotypic heterogeneity of GM1-gangliosidosis, see type I (230500). Description GM1-gangliosidosis type III is an autosomal recessive lysosomal storage disorder characterized by neurodegeneration and mild skeletal changes. Age at onset ranges from 3 to 30 years. The disorder is less severe than GM1-gangliosidosis types I and II (230600). Type III shows extreme clinical variability, with some patients having only focal neurologic signs, such as dystonia, and others having more severe involvement with extrapyramidal signs and mental retardation.
GM1 gangliosidosis is a rare lysosomal storage disorder characterized biochemically by deficient beta-galactosidase activity and clinically by a wide range of variable neurovisceral, ophthalmological and dysmorphic features. Epidemiology The disorder is panethnic but worldwide prevalence is not known. Prevalence at birth is estimated to be approximately 1:100,000 to 200,000 live births. High prevalence has been found in Malta and Brazil, and in the Cypriot and Roma populations. Clinical description There are three types of GM1 gangliosidosis based on the age of onset: a severe rapidly progressive infantile form with onset before six months of age (type 1 GM1 gangliosidosis), a late infantile or juvenile form with onset between seven months and 3 years of age with delayed motor and cognitive development (type 2 GM1 gangliosidosis), and an adult, chronic form with late onset between 3 and 30 years of age (type 3 GM1 gangliosidosis) characterized primarily by generalized dystonia (see these terms).
GM1 gangliosidosis is an inherited lysosomal storage disorder that progressively destroys nerve cells (neurons) in the brain and spinal cord. The condition may be classified into three major types based on the general age that signs and symptoms first appear: classic infantile ( type 1 ); juvenile ( type 2 ); and adult onset or chronic ( type 3 ). Although the types differ in severity, their features may overlap significantly. GM1 gangliosidosis is caused by mutations in the GLB1 gene and is inherited in an autosomal recessive manner. Treatment is currently symptomatic and supportive.
Summary This overview focuses on the clinical features and molecular genetics of common syndromic and nonsyndromic types of hereditary hearing loss. ... In the presence of normal audiometry associated with progressive loss of speech and temporal lobe seizures, the diagnosis of Landau-Kleffner syndrome should be considered. Delayed speech suggesting possible hearing loss can also be seen in young children with autism spectrum disorder or specific speech and language disorders. ... Additionally, thyroid function should be followed if the diagnosis is consistent with Pendred syndrome. Agents/Circumstances to Avoid Noise exposure is a well-recognized environmental cause of hearing loss.
'Autism spectrum disorder,' sometimes referred to as ASD, is a broader phenotype encompassing the less severe disorders Asperger syndrome (see ASPG1; 608638) and pervasive developmental disorder, not otherwise specified (PDD-NOS). ... Mental retardation coexists in approximately two-thirds of individuals with ASD, except for Asperger syndrome, in which mental retardation is conspicuously absent (Jones et al., 2008). ... INHERITANCE - X-linked recessive HEAD & NECK Eyes - Visual problems, mild Mouth - Orofacial hypotonia MUSCLE, SOFT TISSUES - Hypotonia (in some patients) NEUROLOGIC Central Nervous System - Global development delay - Intellectual disability - Learning disability - Motor tics Behavioral Psychiatric Manifestations - Autism spectrum disorder - Attention deficit-hyperactivity disorder - Impulsivity - Aggressive behavior MISCELLANEOUS - Onset in infancy - Variable severity - Nonspecific subtle dysmorphic facial features may be present - Most patients have contiguous gene deletion syndrome involving Xp22 MOLECULAR BASIS - Caused by mutation in the patched domain-containing protein 1 gene (PTCHD1, 300828.0001 ) ▲ Close
The family reported by Ventruto et al. (1976) may have had BDC, but Fitch (1980) favored type B as part of a syndrome. Baraitser and Burn (1983) described an affected brother and sister whose Iraqi first-cousin parents were unaffected, which raised the possibility of autosomal recessive inheritance of this phenotype. ... INHERITANCE - Autosomal dominant SKELETAL Limbs - Madelung deformity Hands - Limited flexion in distal interphalangeal joints - Brachydactyly - Disproportionate shortening of 2nd and 3rd fingers - Short 2nd, 3rd middle phalanges - Ulnar deviation of 2nd, 3rd finger - Hypersegmentation of proximal and middle 2nd, 3rd phalanges - Polydactyly - Fifth finger clinodactyly - Short 1st, 4th, 5th metacarpals Feet - Talipes equinovalgus - Talipes equinovarus MISCELLANEOUS - Allelic to Grebe syndrome ( 200700 ), Du Pan syndrome ( 228900 ), and acromesomelic dysplasia, Hunter Thompson type ( 201250 ) MOLECULAR BASIS - Caused by mutations in the growth/differentiation factor-5 gene (GDF5, 601146.0006 ) ▲ Close
The most common presentation of AA amyloidosis is renal in nature, including proteinuria, nephrotic syndrome and progressive development of chronic kidney disease leading to End Stage Kidney Disease (ESKD) and need for renal replacement therapy (e.g. dialysis or kidney transplantation). [3] A natural history study of AA amyloidosis patients reported a number of conditions associated with AA amyloidosis: [1] Autoimmune diseases Rheumatoid arthritis Ankylosing spondylitis Crohn disease and ulcerative colitis Chronic infections Tuberculosis Bronchiectasis Chronic osteomyelitis Autoinflammatory diseases Familial Mediterranean fever (FMF) Muckle–Wells syndrome (MWS) Cancer Hodgkin's lymphoma Renal cell carcinoma Chronic foreign body reaction HIV/AIDS [4] Silicone-induced granulomatous reaction [5] [6] [7] Pathology [ edit ] In a healthy individual, the median plasma concentration of SAA is 3 mg per liter. [8] This can increase to over 2000 mg per liter during an acute phase response and a sustained overproduction of SAA is required for the creation of the AA deposits that define AA amyloidosis. [9] High levels of SAA, however, is not a sufficient condition for the development of systemic AA amyloidosis and it remains unclear what triggers the accumulation of AA. [10] The AA protein is mainly deposited in the liver, spleen and kidney, and AA amyloidosis can lead to nephrotic syndrome and ESRD. [11] [12] Natural history studies show, however, that it is the kidney involvement that drives the progression of the disease.
Amyloidosis is a group of diseases in which a protein, called amyloid, builds up in the body's organs and tissues. Amyloidosis AA is also referred to as Secondary amyloidosis or Inflammatory amyloidosis. This disease is caused by a long-lasting infection or inflammatory disease such as rheumatoid arthritis, familial Mediterranean fever, or osteomyelitis. Infection or inflammation in the body causes an increased amount of a specific protein called serum amyloid A (SAA) protein. In this disease, part of the SAA protein forms deposits called "amyloid fibrils".
Secondary amyloidosis is a form of amyloidosis (see this term), that complicates chronic inflammatory disorders (mainly rheumatoid arthritis, see this term) and is characterized by the aggregation and deposition of amyloid fibrils composed of serum amyloid A protein, an acute phase reactant. Although spleen, suprarenal gland, liver and gut are frequent sites of amyloid deposition, the clinical picture is dominated by renal involvement.
It is a matter of discussion whether CKD-MBD may be considered a real syndrome or not. [3] CKD-MBD broadens the "old" concept of " renal osteodystrophy ", which now should be restricted to describing the bone pathology associated with CKD. [1] [2] Thus, renal osteodystrophy is currently considered one measure of the skeletal component of the systemic disorder of CKD–MBD that is quantifiable by histomorphometry of bone biopsy. [1] [4] New guidelines have been recently released. [5] Contents 1 Pathophysiology 2 Diagnosis 3 Treatment 4 References 5 External links Pathophysiology [ edit ] It is well-known that as kidney function declines, there is a progressive deterioration in mineral homeostasis, with a disruption of normal serum and tissue concentrations of phosphorus and calcium , and changes in circulating levels of hormones . [2] These include parathyroid hormone (PTH), 25-hydroxyvitamin D (25(OH) vitamin D; calcidiol ), 1,25-dihydroxyvitamin D (1,25(OH)2 vitamin D; calcitriol ), and other vitamin D metabolites, fibroblast growth factor 23 (FGF-23), and growth hormone. [2] Beginning in CKD stage 3, the ability of the kidneys to appropriately excrete a phosphate load is diminished, leading to hyperphosphatemia , elevated PTH ( secondary hyperparathyroidism ), and decreased 1,25(OH)2 vitamin D with associated elevations in the levels of FGF-23. [2] The conversion of 25(OH) vitamin D to 1,25(OH)2 vitamin D is impaired, reducing intestinal calcium absorption and increasing PTH. [2] The kidney fails to respond adequately to PTH, which normally promotes phosphaturia and calcium reabsorption, or to FGF-23, which also enhances phosphate excretion. [2] In addition, there is evidence at the tissue level of a downregulation of vitamin D receptor and of resistance to the actions of PTH. ... The principal conclusion was that the term CKD–Mineral and Bone Disorder (CKD–MBD) should now be used to describe the broader clinical syndrome encompassing mineral, bone, and calcific cardiovascular abnormalities that develop as a complication of CKD . [1] [2] Diagnosis [ edit ] This section is empty. ... "Is chronic kidney disease-mineral bone disorder (CKD-MBD) really a syndrome?" . Nephrol Dial Transplant . 29 (10): 1815–20. doi : 10.1093/ndt/gft514 .
A rare, congenital, esophageal malformation characterized by the presence of an abnormal connection between the esophagus and the trachea (typically occuring in the lower cervical or upper thoracic area and taking an oblique path upward to trachea), without concomitant esophageal atresia. Depending on the size of the lumen, presentation varies from neonatal episodes of choking and cyanosis on feeding to subtle symptoms of wheezing and recurrent respiratory infections in childhood or early adulthood.
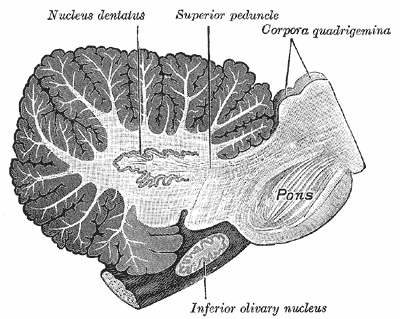
Specialty Neurology Olivopontocerebellar atrophy ( OPCA ) is the degeneration of neurons in specific areas of the brain – the cerebellum , pons , and inferior olivary nucleus . [2] OPCA is present in several neurodegenerative syndromes, including inherited and non-inherited forms of ataxia (such as the hereditary spinocerebellar ataxia known as Machado–Joseph disease ) and multiple system atrophy (MSA) , with which it is primarily associated. [2] OPCA may also be found in the brains of individuals with prion disorders and inherited metabolic diseases . ... Psychiat. 13: 684-702, 1923. ^ MeSH Result ^ https://rarediseases.info.nih.gov/diseases/7250/olivopontocerebellar-atrophy External links [ edit ] Classification D ICD - 10 : G23.8 ICD - 9-CM : 333.0 MeSH : D009849 DiseasesDB : 2012 External resources MedlinePlus : 000758 eMedicine : neuro/282 Olivopontocerebellar atrophy at NINDS v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis
Olivopontocerebellar atrophy (OPCA) is a term used for a progressive condition characterized by the degeneration of nerve cells (neurons) in specific areas of the brain. OPCA can be viewed as a finding of several diseases, and indicates a form of progressive ataxia (abnormal or uncontrolled movements) distinguished by characteristic findings in brain imaging studies and at autopsy (pontine flattening and cerebellar atrophy). It was traditionally divided in hereditary or genetic OPCA and sporadic OPCA. Currently, most of the major forms of hereditary OPCA refer to disorders that overlap with spinocerebellar ataxia (SCA), which is a neurological disorder characterized by ataxia. The sporadic forms are considered now to be a form of multiple system atrophy (MSA).