WH can appear as part of several syndromes, such as Naxos disease (601214) and cardiofaciocutaneous syndrome (115150) (summary by Petukhova et al., 2009).
A number sign (#) is used with this entry because of evidence that autosomal recessive woolly hair-3 (ARWH3) is caused by homozygous mutation in the KRT25 gene (616646) on chromosome 17q21. For a general phenotypic description and a discussion of genetic heterogeneity of autosomal recessive woolly hair, see 278150. Clinical Features Ansar et al. (2015) studied 2 unrelated Pakistani families segregating autosomal recessive woolly hair. In the first family (AP188), 4 individuals from 3 sibships were affected, including 2 brothers and 2 female cousins. Initially, all 4 had soft, short, and sparse hair on the scalp, but at around 4 years of age, they developed tightly curled hair.
A number sign (#) is used with this entry because of evidence that hypotrichosis-13 (HYPT13) is caused by heterozygous mutation in the KRT71 gene (608245) on chromosome 12q13. One such family has been reported. For a discussion of genetic heterogeneity of hypotrichosis, see HYPT1 (605389). Clinical Features Fujimoto et al. (2012) reported a 3-generation Japanese family with woolly hair/hypotrichosis. The proband was a 5-year-old girl who had had tightly curled scalp hair since birth. The scalp hairs were short and stopped growing at a few inches, although there was no apparent hair shaft fragility; no follicular papules were present.
In general, mutations in FGFR1 and FGFR2 cause most of the syndromes involving craniosynostosis, whereas the dwarfing syndromes are largely associated with FGFR3 mutations.
Osteoglophonic dysplasia is a condition characterized by abnormal bone growth that leads to severe head and face (craniofacial) abnormalities, dwarfism, and other features. The term osteoglophonic refers to the bones (osteo-) having distinctive hollowed out (-glophonic) areas that appear as holes on x-ray images. Premature fusion of certain bones in the skull (craniosynostosis) typically occurs in osteoglophonic dysplasia. The craniosynostosis associated with this disorder may give the head a tall appearance, often referred to in the medical literature as a tower-shaped skull , or a relatively mild version of a deformity called a cloverleaf skull . Characteristic facial features in people with osteoglophonic dysplasia include a prominent forehead (frontal bossing ), widely spaced eyes (hypertelorism ), flattening of the bridge of the nose and of the middle of the face (midface hypoplasia ), a large tongue (macroglossia ), a protruding jaw (prognathism ), and a short neck .
A rare disorder characterized by dwarfism, severe craniofacial abnormalities and multiple unerupted teeth. Epidemiology Less than ten cases have been reported so far. Clinical description Main clinical features include craniosynostosis, acrocephaly, a prominent forehead, depressed nasal bridge, hypertelorism, midface hypoplasia, macroglossia, unerupted teeth, short neck, short and bowed limbs, short and broad hands and fingers, and flat feet. The main radiographic features are craniostenosis, fibrous dysplasia, metaphyseal lucencies and platyspondyly. Intelligence is usually normal. Etiology Osteoglosphonic dysplasia (OGD) is caused by mutations in the FGFR1 gene (8p11.2-p11.1). Genetic counseling OGD is transmitted in an autosomal dominant manner.
Mutation in the CDH23 gene can also cause the more severe Usher syndrome 1D (USH1D; 601067). Clinical Features Chaib et al. (1996) described a consanguineous Sunni family with profound prelingual sensorineural hearing impairment living in an isolated village in Syria. ... INHERITANCE - Autosomal recessive HEAD & NECK Ears - Hearing loss, profound prelingual sensorineural Eyes - No retinitis pigmentosa MISCELLANEOUS - Allelic to Usher syndrome, type ID ( 601067 ) MOLECULAR BASIS - Caused by mutation in the cadherin 23 gene (CDH23, 605516.0005 ) ▲ Close
Several other signs and symptoms of the condition are related to the excess IgM, which can thicken blood and impair circulation, causing a condition known as hyperviscosity syndrome. Features related to hyperviscosity syndrome include bleeding in the nose or mouth, blurring or loss of vision, headache, dizziness, and difficulty coordinating movements (ataxia).
Waldenstrom macroglobulinemia is a chronic, slow-growing lymphoproliferative disorder . It usually affects older adults and is primarily found in the bone marrow, although lymph nodes and the spleen may be involved. Affected individuals have a high level of an antibody called immunoglobulin M (IgM) in their blood, which can cause thickening of the blood (hyperviscosity). Although some individuals initially do not have symptoms and are diagnosed from routine blood work, common symptoms may include weakness, appetite loss and weight loss. Other symptoms may include peripheral neuropathy, fever, Raynaud's phenomenon , and mental status changes.
Paragangliomas and pheochromocytomas can occur in individuals with other inherited disorders, such as von Hippel-Lindau syndrome, Carney-Stratakis syndrome, and certain types of multiple endocrine neoplasia.
The triad of gastric leiomyosarcoma, pulmonary chondroma, and extraadrenal paraganglioma constitutes a syndrome that occurs mainly in young women and is known as the Carney triad (604287). This triad is not to be confused with the other Carney syndrome of myxoma, spotty pigmentation, and endocrinopathy (160980). ... Nissenblatt (1978) reported a young woman with hypoplastic right heart syndrome who developed a carotid body tumor at age 28 years. ... In 16 patients who had other extraadrenal paragangliomas, suggesting a multiple primary tumor syndrome, the diagnosis occurred earlier (mean, 35.4 years; p less than 0.01). ... Parkin (1981) reported 2 unrelated families with a hereditary syndrome of pheochromocytoma associated with multiple glomus tumors of the head and neck.
While most are single, rare multiple cases occur (usually in a hereditary syndrome). Paragangliomas are described by their site of origin and are often given special names: Head and neck paraganglioma (HNPGL): There are various types of head and neck paraganglioma; they may have specialized names depending on the precise location. [8] Carotid paraganglioma ( carotid body tumor): Is the most common of the head and neck paragangliomas. ... "Metabolic Drivers in Hereditary Cancer Syndromes" . Annual Review of Cancer Biology . 4 : 77–97. doi : 10.1146/annurev-cancerbio-030419-033612 . ^ Feky, Mostafa Mahmoud El. ... External links [ edit ] GeneReviews/NCBI/NIH/UW entry on Hereditary Paraganglioma-Pheochromocytoma Syndromes k Classification D ICD - 10 : C75.4 , C75.5 , D35.5 , D35.6 , D44.6 , D44.7 ICD - 9-CM : 194.5 , 194.6 , 227.5 , 227.6 , 237.3 ICD-O : M8680/0 - M8693/9 MeSH : D010235 DiseasesDB : 33480 SNOMED CT : 302833002 External resources eMedicine : med/2994 v t e Tumours of endocrine glands Pancreas Pancreatic cancer Pancreatic neuroendocrine tumor α : Glucagonoma β : Insulinoma δ : Somatostatinoma G : Gastrinoma VIPoma Pituitary Pituitary adenoma : Prolactinoma ACTH-secreting pituitary adenoma GH-secreting pituitary adenoma Craniopharyngioma Pituicytoma Thyroid Thyroid cancer (malignant): epithelial-cell carcinoma Papillary Follicular / Hurthle cell Parafollicular cell Medullary Anaplastic Lymphoma Squamous-cell carcinoma Benign Thyroid adenoma Struma ovarii Adrenal tumor Cortex Adrenocortical adenoma Adrenocortical carcinoma Medulla Pheochromocytoma Neuroblastoma Paraganglioma Parathyroid Parathyroid neoplasm Adenoma Carcinoma Pineal gland Pinealoma Pinealoblastoma Pineocytoma MEN 1 2A 2B v t e Tumours of the nervous system Endocrine Sellar : Craniopharyngioma Pituicytoma Other: Pinealoma CNS Neuroepithelial ( brain tumors , spinal tumors ) Glioma Astrocyte Astrocytoma Pilocytic astrocytoma Pleomorphic xanthoastrocytoma Subependymal giant cell astrocytoma Fibrillary astrocytoma Anaplastic astrocytoma Glioblastoma multiforme Oligodendrocyte Oligodendroglioma Anaplastic oligodendroglioma Ependyma Ependymoma Subependymoma Choroid plexus Choroid plexus tumor Choroid plexus papilloma Choroid plexus carcinoma Multiple/unknown Oligoastrocytoma Gliomatosis cerebri Gliosarcoma Mature neuron Ganglioneuroma : Ganglioglioma Retinoblastoma Neurocytoma Dysembryoplastic neuroepithelial tumour Lhermitte–Duclos disease PNET Neuroblastoma Esthesioneuroblastoma Ganglioneuroblastoma Medulloblastoma Atypical teratoid rhabdoid tumor Primitive Medulloepithelioma Meninges Meningioma Hemangiopericytoma Hematopoietic Primary central nervous system lymphoma PNS : Nerve sheath tumor Cranial and paraspinal nerves Neurofibroma Neurofibromatosis Neurilemmoma / Schwannoma Acoustic neuroma Malignant peripheral nerve sheath tumor Other WHO classification of the tumors of the central nervous system Note: Not all brain tumors are of nervous tissue, and not all nervous tissue tumors are in the brain (see brain metastasis ).
Gangliocytic paraganglioma Micrograph of a gangliocytic paraganglioma. H&E stain . Pronunciation GP Specialty Pathology A gangliocytic paraganglioma is a rare tumour that is typically found in the duodenum and consists of three components: (1) ganglion cells, (2) epithelioid cells ( paraganglioma -like) and, (3) spindle cells ( schwannoma -like). [1] Contents 1 Symptoms and signs 2 Pathology 3 See also 4 References Symptoms and signs [ edit ] The most common presentation is gastrointestinal bleed (~45% of cases), followed by abdominal pain (~43% of cases) and anemia (~15% of cases). [2] Pathology [ edit ] GP consist of three components (1) ganglion cells , (2) epithelioid cells (neuroendocrine-like), and (3) spindle cells ( schwannoma -like). The microscopic differential diagnosis includes poorly differentiated carcinoma, neuroendocrine tumour and paraganglioma . [1] GPs may be sporadic or arise in the context neurofibromatosis type 1 . Intermed. mag. Intermed. mag. Very high mag. Very high mag. See also [ edit ] Schwannoma Paraganglioma References [ edit ] ^ a b Wong, A.; Miller, AR.; Metter, J.; Thomas, CR. (Mar 2005). "Locally advanced duodenal gangliocytic paraganglioma treated with adjuvant radiation therapy: case report and review of the literature" .
At one point, only 45 families worldwide were known to possess this genetic trait. [6] It is now more widely recognized, although the precise number of people affected with this form of DI is unknown at the present time. [ citation needed ] There is also an X-linked familial form.Wolfram Syndrome (also called DIDMOAD) is characterised by DI, diabetes mellitus nerve deafness and optic atrophy. [ citation needed ] Diagnosis [ edit ] In many respects, the diagnosis of central diabetes insipidus begins as a diagnosis of exclusion. ... Cite journal requires |journal= ( help ) http://diabetesinsipidus.org/4di_familial.htm Archived 2009-04-29 at the Wayback Machine External links [ edit ] Classification D ICD - 10 : E23.2 ICD - 9-CM : 253.5 MeSH : D020790 v t e Hypothalamic disease Gonadotropin Kallmann syndrome Adiposogenital dystrophy CRH Tertiary adrenal insufficiency Vasopressin Neurogenic diabetes insipidus General Hypothalamic hamartoma
A number sign (#) is used with this entry because neurohypophyseal diabetes insipidus is caused by heterozygous mutation in the arginine vasopressin gene (AVP; 192340) on chromosome 20p13. An X-linked form of neurohypophyseal diabetes insipidus (304900) has been suggested, but the evidence is weak. Description Neurohypophyseal diabetes insipidus is an autosomal dominant disorder of free water conservation characterized by childhood onset of polyuria and polydipsia. Affected individuals are apparently normal at birth, but characteristically develop symptoms of vasopressin deficiency during childhood (summary by Wahlstrom et al., 2004). Clinical Features Normally the posterior pituitary hormones, antidiuretic hormone and oxytocin, are synthesized in the supraoptic and paraventricular nuclei of the hypothalamus and transported within axons, possibly in a biologically inactive, bound form, to the posterior lobe of the pituitary where they are stored.
Recently, the dosage of aquaporin 2 (AQP2) has been used in the differential diagnosis of CDI as the failure to increase AQP2 excretion after desmopressin administration indicates a nephrogenic form of diabetes insipidus. Wolfram syndrome (see this term) is another differential diagnosis.
Central diabetes insipidus (DI) is a form of DI that occurs when the body has lower than normal levels of antidiuretic hormone (vasopressin), which is characterized by frequent urination. Diabetes insipidus is subdivided into central and nephrogenic DI . Two other forms are gestational DI and primary polydipsia ( dipsogenic DI ). Central DI results from damage to the pituitary gland , which disrupts the normal storage and release of antidiuretic hormone (ADH). When this hormone reaches the kidneys, it directs them to make less urine. The major symptoms of central diabetes insipidus (DI) include urinating too much (polyuria), getting up at night to urinate (nocturia), and drinking too much liquids (polydipsia).
Neurohypophyseal diabetes insipidus is a disorder of water balance. The body normally balances fluid intake with the excretion of fluid in urine. However, people with neurohypophyseal diabetes insipidus produce too much urine (polyuria), which causes them to be excessively thirsty (polydipsia). Affected people need to urinate frequently, which can disrupt daily activities and sleep. People with neurohypophyseal diabetes insipidus can quickly become dehydrated if they do not drink enough water. Dehydration can lead to constipation and dry skin. If the disorder is not treated, more serious complications of dehydration can occur.
Hereditary central diabetes insipidus is a rare genetic subtype of central diabetes insipidus (CDI, see this term) characterized by polyuria and polydipsia due to a deficiency in vasopressin (AVP) synthesis. Epidemiology The prevalence is unknown. Clinical description Symptoms usually develop between 1 and 6 years of age but onset in the neonatal period or in elderly patients has been described. They include polyuria, polydipsia and nocturia (often manifesting as enuresis in children). Additional symptoms seen in children can include: lethargy, irritability, growth retardation, weight loss, fever, vomiting or diarrhea. In the autosomal recessive form, symptoms are secondary to reduced biological activity of mutant AVP; heterozygous carriers have subclinical manifestations or are asymptomatic.
In addition to the X-linked forms of diabetes insipidus, autosomal dominant forms also exist. Forssman (1955) had 5 families: 2 probably autosomal and 3 X-linked. Of the 3 X-linked families, 1 was of the pitressin-resistant type, whereas the other 2 families were susceptible. The latter 2 families presumably represent the neurohypophyseal type. Green et al. (1967) reported a family with diabetes insipidus in a dominant pattern, either X-linked or autosomal. Autopsy in one of the affected members showed marked reduction of neurons in the supraoptic and paraventricular nuclei of the hypothalamus.
It is suspected that AMC is related to decreased fetal movement during development which can have a variety of different causes, including environmental factors (i.e. maternal illness, limited space), single gene changes (autosomal dominant, autosomal recessive, X-linked), chromosomal abnormalities and various syndromes. Treatment varies based on the signs and symptoms found in each person, but may include physical therapy, removable splints, exercise, and/or surgery.
A group of disorders characterized by congenital limb contractures manifesting as limitation of movement of multiple limb joints at birth that is usually non-progressive and may include muscle weakness and fibrosis. This disorder is always associated with decreased intrauterine fetal movement which leads secondarily to the contractures.
Quercia and Teebi (2002) suggested that inheritance is probably autosomal dominant with variable expression and incomplete penetrance. They concluded that the syndrome includes congenital heart defects.
Besides cystic fibrosis, it is also seen in wasting ( marasmus ) and nephrotic syndrome and also with the use of aspirin and other drugs such as rofecoxib and celecoxib .
A rare skin disease characterized by transient wrinkling of the skin, edema, formation of whitish papules, pruritus, burning sensation, or pain, on the palms and/or soles in response to contact with water. Duration of exposure and water temperature affect the rate of development and intensity of the lesions. The condition is more common in females than in males and frequently occurs in patients with cystic fibrosis.
., kinesigenic (128200); with Lesch-Nyhan syndrome (308000); with Wilson disease (277900); with acanthocytosis (200150) and sometimes with familial basal ganglion calcification (114100).
A rare motor-sensory, axonal form of Guillain-Barré syndrome (GBS). Epidemiology The overall annual incidence of GBS is estimated at between 1/91,000 and 1/55,000.
Multiple cutaneous leiomyoma Other names Hereditary leiomyomatosis and renal cell cancer [1] Specialty Dermatology Multiple cutaneous leiomyomas , also known as Pilar leiomyomas , [2] arise from the arrectores pilorum muscles, and are made up of a poorly circumscribed proliferation of haphazardly arranged smooth muscle fibers located in the dermis that appear to infiltrate the surrounding tissue and may extend into the subcutis. [2] Sometimes associated with uterine leiomyomas (a combination known as multiple cutaneous and uterine leiomyomatosis , MCUL), these lesions may also be a manifestation of the hereditary leiomyomatosis and renal cell cancer syndrome. See also [ edit ] List of cutaneous conditions References [ edit ] ^ "Hereditary leiomyomatosis and renal cell cancer | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov .
., glycogen storage diseases, the atonic-astatic syndrome of Foerster (209100), and the congenital nonprogressive myopathy described by Batten and by Turner (255300).
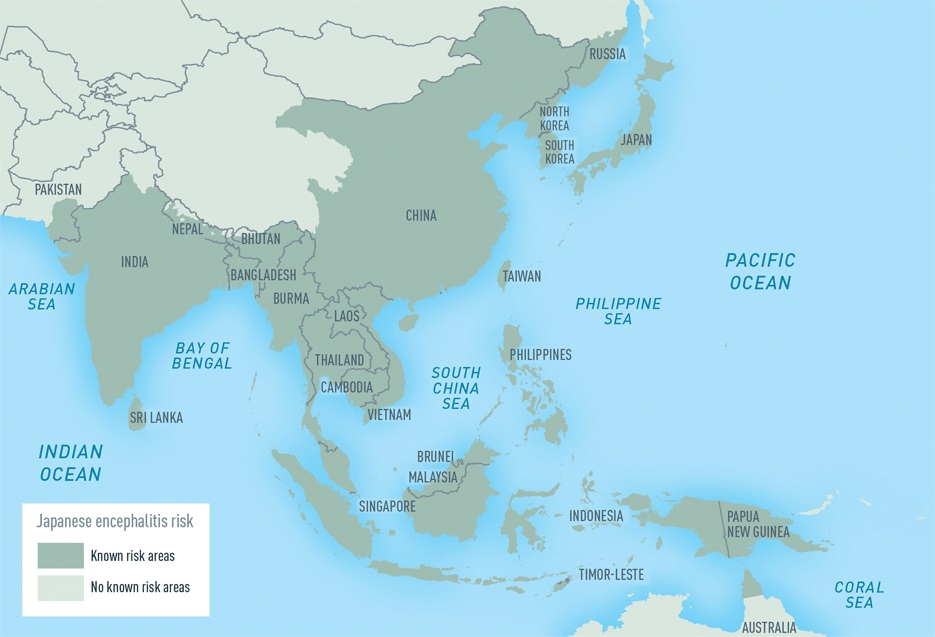
Clinical description After an incubation period of four to 14 days, infected individuals present with signs ranging from moderate with a headache and low-grade fever to more severe infection with high fever, meningeal syndrome (neck stiffness, vomiting), disorientation and sometimes tremors or coma.
Refractory cytopenias with unilineage dysplasia (RCUD) is a frequent low-risk subtype of myelodysplastic syndrome (MDS; see this term) characterized by refractory cytopenias associated with dysplasia limited to one cell lineage.
Congenital radioulnar synostosis is a rare bone disorder that may be isolated or associated with other disorders and that is characterized by failure of segmentation of the radius and ulna during embryological development, causing limited rotational movements of the forearm, which may lead to difficulties with some activities of daily living.
Radioulnar synostosis is a feature of certain chromosome abnormalities, notably the triple X-Y syndrome (XXXY). See pronation-supination of the forearm, impairment of (176800).