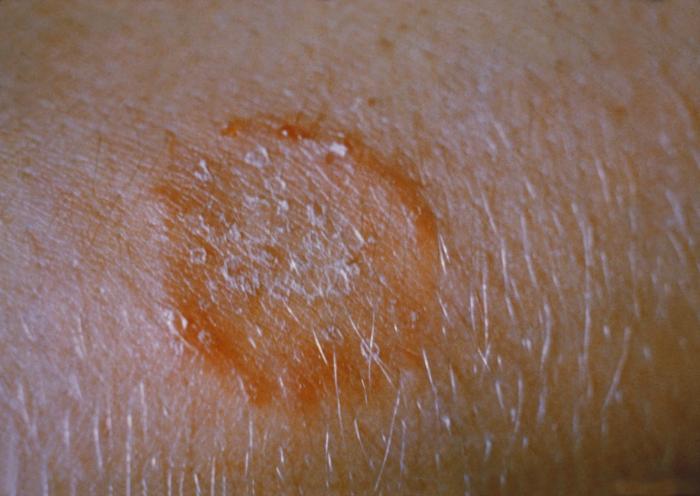
It may result in aesthetic or functional sequelae with variable expression depending on the sites involved (deafness, respiratory or hepatic failure, diabetes insipidus, growth hormone deficiency, and cerebellar syndrome). In adults, the clinical picture is characterised by isolated lung disease, with a strong association with smoking.
. ^ The Writing Group of the Histiocyte Society (1987). "Histiocytosis syndromes in children. Writing Group of the Histiocyte Society". ... Defects in membranous bones, exophthalmos, and diabetes insipidus; an unusual syndrome of dyspituitarism. In: Contributions to medical and biological research, dedicated to Sir William Osler.
Langerhans cell histiocytosis is a disorder in which excess immune system cells called Langerhans cells build up in the body. Langerhans cells, which help regulate the immune system, are normally found throughout the body, especially in the skin , lymph nodes, spleen , lungs, liver , and bone marrow. In Langerhans cell histiocytosis, excess immature Langerhans cells usually form tumors called granulomas. Many researchers now consider Langerhans cell histiocytosis to be a form of cancer, but this classification remains controversial. In approximately 80 percent of affected individuals, one or more granulomas develop in the bones, causing pain and swelling.
Clinical manifestations range from a spontaneously healing isolated osteolytic lesion to a lymphoma-like syndrome with fatal multiorgan failure, in the absence of any cellular evidence of malignancy. ... Egeler and D'Angio (1995) presented a classification of histiocytosis syndromes in children: class I, Langerhans cell histiocytosis (LCH); class II, histiocytosis of mononuclear macrophages other than Langerhans cells, including familial hemophagocytic lymphohistiocytosis (267700); and class III, malignant histiocytic disorders, including histiocytic lymphoma.
Non-X histiocytosis Specialty Dermatology Non-X histiocytoses are a clinically well-defined group of cutaneous syndromes characterized by infiltrates of monocytes / macrophages , as opposed to X-type histiocytoses in which the infiltrates contain Langerhans cells . [1] : 714 Conditions included in this group are: [1] : 714–20 Juvenile xanthogranuloma Benign cephalic histiocytosis Generalized eruptive histiocytoma Xanthoma disseminatum Progressive nodular histiocytosis Papular xanthoma Hereditary progressive mucinous histiocytosis Reticulohistiocytosis Indeterminate cell histiocytosis Sea-blue histiocytosis Erdheim–Chester disease See also [ edit ] X-type histiocytosis Histiocytosis References [ edit ] ^ a b James, William D.; Berger, Timothy G.; et al. (2006).
Pityriasis lichenoides (PL) is a skin condition characterized by small, raised pink spots that tend to come together in groups. It is not contagious. There are two main types of PL: an acute form called pityriasis lichenoides et varioliformis acuta (PLEVA), and a milder, longer-lasting form called pityriasis lichenoides chronica (PLC). There is also a rare, severe variant of PLEVA called febrile ulceronecrotic PLEVA, associated with high fever and complications that may affect other body systems. In both types of PL, spots usually occur on the trunk, buttox, arms and legs. PLEVA begins abruptly and may cause itching or burning. PLC may develop over days, is less irritating, and lasts longer than PLEVA.
Adults who've had repair surgery for a congenital heart defect also have an increased risk of sudden cardiac arrest. Long QT syndrome (LQTS) and other heart signaling problems. Conditions such as long QT syndrome and Brugada syndrome cause the heart to beat in an unorganized way. ... Genetic tests can be done to see if you have long QT syndrome, a common cause of sudden cardiac death.
Their affected mother had a progressive interictal cerebellar syndrome with ataxia and dysarthria. The report indicated that nongenetic factors play a role in the severity of EA1. ... Affected persons probably have an increased frequency of muscle cramps ('night cramps'). A syndrome of continuous muscle fiber activity was described by Ashizawa et al. (1983). ... McGuire et al. (1984) described the syndrome of continuous muscle fiber activity in a 3-year-old boy and his 28-year-old mother. ... Jen et al. (2007) provided a detailed review of the pathophysiology and molecular genetics of known episodic ataxia syndromes. Animal Model Using homologous recombination, Herson et al. (2003) introduced the Kcna1 val408-to-ala mutation (V408A; 176260.0001) into mice.
Hereditary continuous muscle fiber activity is a rare, non-dystrophic myopathy characterized by generalized myokymia and increased muscle tone associated with delayed motor milestones, leg stiffness, spastic gait, hyperreflexia and Babinski sign. Symptoms may be worsened by febrile illness or anesthesia.
While UCD is readily treatable with surgery, HHV-8-associated MCD, like iMCD, is treated with medications as surgery is ineffective. [6] TAFRO Syndrome: a constellation of clinical symptoms reported in some iMCD and HHV-8 associated MCD patients. ... All cases of HHV-8-associated MCD are thought to demonstrate plasmablastic features—similar to plasmacytic features, but with plasmablasts present. [10] The clinical utility of subtyping Castleman disease by histologic features is uncertain, as histologic subtypes do not consistently predict disease severity or treatment response. [10] Staining with latency-associated nuclear antigen (LANA-1) , a marker for HHV-8 infection, is positive only in HHV-8-associated MCD. [12] Diseases other than Castleman disease can present with similar histologic findings in lymph node tissue, including: [10] Infectious causes : Epstein-Barr virus , human immunodeficiency virus , tuberculosis Autoimmune diseases : Systemic lupus erythematosus , rheumatoid arthritis Lymphoproliferative disorders : lymphoma , autoimmune lymphoproliferative syndrome History [ edit ] Unicentric Castleman disease was first described in a case series by Benjamin Castleman in 1956. [13] By 1984, a number of case reports had been published describing a multicentric variant of the disease and with some reports describing an association with Kaposi's sarcoma . [14] In 1995, the association between HHV-8 and Castleman disease was described in patients with HIV . [15] Formal diagnostic criteria and definition of the disease was established in 2016, which will allow for better understanding and the ability to appropriately track and research CD. ... "Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease" . ... "Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease" .
Progranulin is associated with tumorgenesis when overproduced, however the mutations seen in FTLD-TDP43 produce a haploinsufficiency, meaning that because one of the two alleles is damaged, only half as much Progranulin is produced. [5] Mutations in the CHMP2B gene are associated with a rare behavioural syndrome akin to bvFTLD (mainly in a large Jutland cohort), presenting with a tau negative, TDP-43 negative, FUS negative, Ubiquitin positive pathology. ... Further reading [ edit ] Hodges, John R. The Frontotemporal Dementia Syndromes. Cambridge University P ress. 2007 ISBN 978-0-521-85477-1 OMIM entries on FRONTOTEMPORAL DEMENTIA AND/OR AMYOTROPHIC LATERAL SCLEROSIS as well as C9ORF72 GeneReviews/NCBI/NIH/UW entry on Amyotrophic Lateral Sclerosis Overview External links [ edit ] Classification D ICD - 10 : G31.0 OMIM : 600274 MeSH : D003704 DiseasesDB : 10034 v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis
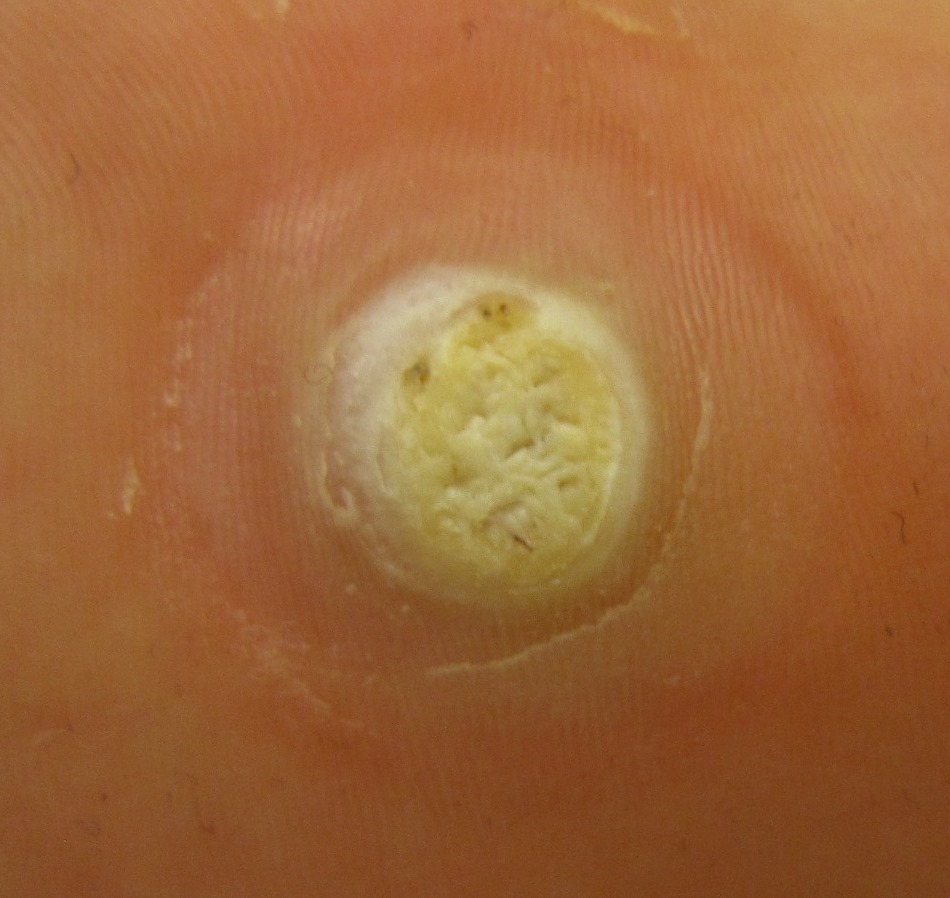
Overview Plantar warts are small, rough growths on the feet. They usually show up on the balls and heels of the feet, the areas that bear the most pressure. This pressure may also cause a wart to grow inward beneath a hard, thick layer of skin (callus). Plantar warts are caused by HPV. This virus enters through tiny cuts or breaks on the bottom of the feet. Most plantar warts aren't a serious health concern and often go away without treatment, especially in children under 12. To get rid of them sooner, you can try self-care treatments or see your health care provider.
Type 1 is associated with pancreatitis, Sjogren syndrome, Primary sclerosing cholangitis and Inflammatory bowel disease. ... AIP occurring in association with an autoimmune disorder has been referred to as "secondary" or "syndromic" AIP. AIP does not affect long-term survival. [3] Contents 1 Signs and symptoms 2 Histopathology 3 Diagnosis 3.1 Criteria 3.2 Radiologic features 4 Treatment 5 Controversies in nomenclature 6 Prognosis 7 Epidemiology 8 References 9 External links Signs and symptoms [ edit ] Autoimmune pancreatitis may cause a variety of symptoms and signs, which include pancreatic and biliary (bile duct) manifestations, as well as systemic effects of the disease. ... Additional manifestations include inflammation in the salivary glands ( Sjögren syndrome ), in the lungs resulting in scarring ( pulmonary fibrosis ) and nodules , scarring within the chest cavity ( mediastinal fibrosis ) or in the anatomic space behind the abdomen ( retroperitoneal fibrosis ) and inflammation in the kidneys ( tubulointerstitial nephritis ). [4] AIP is characterized by the following features: Scleral Icterus (yellow eyes), jaundice (yellow skin) which is usually painless, usually without acute attacks of pancreatitis . ... There are also a large number of case reports employing descriptive terminology such as pancreatitis associated with Sjögren’s syndrome , primary sclerosing cholangitis , or inflammatory bowel disease .
Autoimmune pancreatitis affects the pancreas, a gland behind the stomach and in front of the spine, and can also affect the bile ducts, salivary glands, kidneys, and lymph nodes. It is thought to occur when the immune system mistakenly begins to attack these healthy body tissues, glands, and organs. Common signs and symptoms include painless jaundice, weight loss, and noncancerous masses in the pancreas and other organs. Treatment often involves corticosteroids. The condition may recur following treatment, and require additional therapy.
Overview Autoimmune pancreatitis (AIP) is a chronic inflammation that is thought to be caused by the body's immune system attacking the pancreas and that responds to steroid therapy. Two subtypes of AIP are now recognized, type 1 and type 2. Type 1 AIP is the pancreatic manifestation of a disease called IgG4-related disease (IgG4-RD). This disease often affects multiple organs including the pancreas, bile ducts in the liver, salivary glands, kidneys and lymph nodes. Type 2 AIP seems to affect only the pancreas, although about one-third of people with type 2 AIP have associated inflammatory bowel disease. Type 1 AIP can be mistakenly diagnosed as pancreatic cancer. The two conditions have overlapping signs and symptoms, but very different treatments, so it is very important to distinguish one from the other.
A rare pancreatic disease characterized by chronic non-alcoholic pancreatitis that presents with abdominal pain, steatorrhea, obstructive jaundice and responds well to steroid therapy and is seen in two subforms: type which affects elderly males, involves other organs and has increased immunoglobin G4 (IgG4) levels and type 2 which affects both sexes equally but presents at a younger age and has no other organ involvement or increased IgG4 levels.
Specialty Oncology , Gastroenterology Usual onset 38 years of age (median) Causes Mutation of the E-cadherin gene (CDH1) Risk factors Stomach cancer Treatment Total gastrectomy Frequency 1-3% of gastric cancers Hereditary diffuse gastric cancer (HDGC) is an inherited genetic syndrome most often caused by an inactivating mutation in the E-cadherin gene (CDH1) located on chromosome 16 . [1] Individuals who inherit an inactive copy of the CDH1 gene are at significantly elevated risk for developing stomach cancer . ... An estimated 1-3% of gastric cancers are associated with hereditary cancer syndromes. HDGC is the most common hereditary cancer syndrome of the stomach. [4] HDGC was originally discovered through studies of Maori families in New Zealand that were noted to have increased incidences of gastric cancer. [2] Detection of CDH1 mutations causing HDGC is highest in countries with low incidences of gastric cancer, such as the United States and Canada. ... "Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond" . JAMA Oncology . 1 (1): 23–32. doi : 10.1001/jamaoncol.2014.168 .
Description Hereditary diffuse gastric cancer is an autosomal dominant cancer predisposition syndrome. Heterozygous CDH1 mutation carriers have a 70 to 80% lifetime risk of developing diffuse gastric cancer. ... Inheritance Hereditary diffuse gastric cancer is an autosomal dominant cancer predisposition syndrome. Heterozygous CDH1 mutation carriers have a 70 to 80% lifetime risk of developing gastric cancer.
Hereditary diffuse gastric cancer (HDGC) leads to an increased risk (predisposition) of developing a specific form of stomach cancer called diffuse gastric cancer . Women with HDGC also have an increased risk for lobular breast cancer . Cancers associated with HDGC generally occur at earlier ages than those seen in people who do not have a hereditary predisposition to cancer. HDGC is caused by genetic variants in the CDH1 gene and the CTNNA1 gene. It is inherited in an autosomal dominant pattern. Diagnosis of HDGC is based on the symptoms, family history, and may be confirmed by the results of genetic testing.
Hereditary diffuse gastric cancer (HDGC) is an inherited disorder that greatly increases the chance of developing a form of stomach(gastric) cancer. In this form, known as diffuse gastric cancer, there is no solid tumor. Instead cancerous (malignant) cells multiply underneath the stomach lining , making the lining thick and rigid. The invasive nature of this type of cancer makes it highly likely that these cancer cells will spread (metastasize ) to other tissues, such as the liver or nearby bones. Symptoms of diffuse gastric cancer occur late in the disease and can include stomach pain, nausea, vomiting, difficulty swallowing (dysphagia), decreased appetite, and weight loss.
Hereditary diffuse gastric cancer is a rare epithelial tumor of the stomach, characterized by the development of diffuse (signet ring cell) gastric cancer at a young age, associated with germline heterozygous mutations of CDH1 , MAP3K6 and CTNNA1 genes. In early stages it presents with non-specific and vague symptoms, in advanced stages it may cause nausea and vomiting, dysphagia, loss of appetite, abdominal mass or weight loss. Women have an increased risk of lobular breast cancer as well.
Summary Clinical characteristics. The classic phenotype of megalencephalic leukoencephalopathy with subcortical cysts (MLC) is characterized by early-onset macrocephaly, often in combination with mild gross motor developmental delay and seizures; gradual onset of ataxia, spasticity, and sometimes extrapyramidal findings; and usually late onset of mild mental deterioration. Macrocephaly, observed in virtually all individuals, may be present at birth but more frequently develops during the first year of life. The degree of macrocephaly is variable and can be as great as 4 to 6 SD above the mean in some individuals. After the first year of life, head growth rate normalizes and growth follows a line parallel to and usually several centimeters above the 98th centile. Initial mental and motor development is normal in most individuals. Walking is often unstable, followed by ataxia of the trunk and extremities, then minor signs of pyramidal dysfunction and brisk deep-tendon stretch reflexes.
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a condition that affects brain development and function. Individuals with this condition have an enlarged brain ( megalencephaly ) and an abnormality of the white matter in the brain ( leukoencephalopathy ). White matter consists of nerve fibers covered by a fatty substance called myelin that promotes the rapid transmission of nerve impulses. In MLC, the myelin is swollen and contains numerous fluid-filled pockets (vacuoles). Over time, the swelling decreases and the myelin begins to waste away (atrophy).
A number sign (#) is used with this entry because remitting megalencephalic leukoencephalopathy with subcortical cysts-2B (MLC2B) is caused by heterozygous mutation in the HEPACAM gene (611642) on chromosome 11q24. Description Autosomal dominant remitting MLC2B is characterized by infantile-onset of macrocephaly and mildly delayed motor development associated with white matter abnormalities on brain MRI that improve with age. As children, some patients have mild residual hypotonia or clumsiness, but otherwise have no residual motor abnormalities. About 40% of patients have mental retardation (summary by van der Knaap et al., 2010 and Lopez-Hernandez et al., 2011). Homozygous or compound heterozygous mutations in the HEPACAM gene can cause a more severe and progressive disorder associated with ataxia, spasticity, and mental retardation (MLC2A; 613925).
Megalencephalic leukoencephalopathy with subcortical cysts is a progressive condition that affects brain development and function. Individuals with this condition typically have an enlarged brain (megalencephaly) that is evident at birth or within the first year of life. Megalencephaly leads to an increase in the size of the head (macrocephaly ). Affected people also have leukoencephalopathy, an abnormality of the brain's white matter. White matter consists of nerve fibers covered by a fatty substance called myelin .
A number sign (#) is used with this entry because autosomal recessive megalencephalic leukoencephalopathy with subcortical cysts-2A (MLC2A) is caused by homozygous or compound heterozygous mutation in the HEPACAM gene (611642) on chromosome 11q24. For a discussion of genetic heterogeneity of megalencephalic leukoencephalopathy with subcortical cysts, see MLC1 (604004). Description Megalencephalic leukoencephalopathy with subcortical cysts-2A is an autosomal recessive neurodegenerative disorder characterized by infantile-onset macrocephaly and later onset of motor deterioration, with ataxia and spasticity, seizures, and cognitive decline of variable severity. Brain MRI shows typical white matter abnormalities, including swelling of the cerebral white matter and subcortical cysts, in all stages of the disease (summary by Lopez-Hernandez et al., 2011). Heterozygous mutations in the HEPACAM gene can cause a similar, but less severe disorder that shows improvement of MRI changes with age (MLC2B; 613926).
Clinical Features Van der Knaap et al. (1995) described a syndrome of cerebral leukoencephalopathy and megalencephaly with infantile onset in 8 children, including 2 sibs.
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a form of leukodystrophy that is characterized by infantile-onset macrocephaly, often with mild neurologic signs at presentation (such as mild motor delay), which worsen with time, leading to poor ambulation, falls, ataxia, spasticity, increasing seizures and cognitive decline. Brain magnetic resonance imaging reveals diffusely abnormal and mildly swollen white matter as well as subcortical cysts in the anterior temporal and frontoparietal regions.
Clinical Features Guran et al. (2019) reported 4 unrelated 46,XY females from consanguineous Turkish families with syndromic complete gonadal dysgenesis. The genital phenotype was female with hypoplastic labia majora; uterus was present in 3 patients and a primitive bicornuate uterus was detected in the fourth patient. ... Molecular Genetics In 4 unrelated 46,XY females from consanguineous Turkish families with syndromic complete gonadal dysgenesis, Guran et al. (2019) identified homozygosity for 3 different missense mutations in the PPP2R3C gene (615902.0001-615902.0003) that segregated with disease in each family and were not found in ethnically matched controls or in public variant databases.
Additionally, the condition is a characteristic feature of certain inherited syndromes such as megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS) or mitochondrial neurogastrointestinal encephalopathy disease (MNGIE disease).
Description Premature ovarian failure-10 (POF10) represents a syndrome characterized by primary amenorrhea, hypergonadotropic ovarian insufficiency, and genomic instability in somatic cells. ... Her 21-year-old 46,XY brother, who had been diagnosed with a 22q11 microdeletion (DiGeorge syndrome; 188400), exhibited normal pubic hair and penile development, but testicular volume was only 3 mL and he was azoospermic with high basal and GnRH-stimulated gonadotropins, consistent with primary testicular failure.
'Autism spectrum disorder,' sometimes referred to as ASD, is a broader phenotype encompassing the less severe disorders Asperger syndrome (see ASPG1; 608638) and pervasive developmental disorder, not otherwise specified (PDD-NOS). ... Mental retardation coexists in approximately two-thirds of individuals with ASD, except for Asperger syndrome, in which mental retardation is conspicuously absent (Jones et al., 2008).