-
Torch Syndrome
Wikipedia
TORCH syndrome Other names TORCH infection TORCH syndrome is a cluster of symptoms caused by congenital infection with toxoplasmosis , rubella , cytomegalovirus , herpes simplex , and other organisms including syphilis , parvovirus , and Varicella zoster . [1] Zika virus is considered the most recent member of TORCH infections. [2] "TORCH" is an acronym for (T)oxoplasmosis, (O)ther Agents, (R)ubella, (C)ytomegalovirus, and (H)erpes Simplex. [3] Contents 1 Signs and symptoms 2 Pathophysiology 3 Diagnosis 4 Prevention 5 Treatment 6 Epidemiology 7 References Signs and symptoms [ edit ] Though caused by different infections, the signs and symptoms of TORCH syndrome are consistent. ... The specific infection may cause additional symptoms. [1] TORCH syndrome may develop before birth, causing stillbirth, in the neonatal period, or later in life. [4] Pathophysiology [ edit ] TORCH syndrome is caused by in utero infection with one of the TORCH agents, disrupting fetal development . [1] Diagnosis [ edit ] Presence of IgM is diagnostic and Persistence of IgG beyond 6–9 months is diagnostic [ citation needed ] Prevention [ edit ] TORCH syndrome can be prevented by treating an infected pregnant person, thereby preventing the infection from affecting the fetus. [4] Treatment [ edit ] The treatment of TORCH syndrome is mainly supportive and depends on the symptoms present; medication is an option for herpes and cytomegalovirus infections. [1] Epidemiology [ edit ] Developing countries are more severely affected by TORCH syndrome. [4] References [ edit ] ^ a b c d "TORCH Syndrome - NORD (National Organization for Rare Disorders)" . ... PMID 28579764 . ^ https://rarediseases.org/rare-diseases/torch-syndrome/ ^ a b c Neu, Natalie; Duchon, Jennifer; Zachariah, Philip (2015-03-01).
-
Microphthalmia–dermal Aplasia–sclerocornea Syndrome
Wikipedia
Microphthalmia–dermal aplasia–sclerocornea syndrome Other names MIDAS syndrome [1] Microphthalmia–dermal aplasia–sclerocornea syndrome is a condition characterized by linear skin lesions . [1] [2] MLS is a rare X-linked dominant male -lethal disease characterized by unilateral or bilateral microphthalmia and linear skin defects in affected females , and in utero lethality for affected males. [3] It can be associated with HCCS , [4] but mutations in the MCCS gene cause microphthalmia with Linear Skin Defects Syndrome. [5] See also [ edit ] List of cutaneous conditions References [ edit ] ^ a b Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... ISBN 978-1-4160-2999-1 . ^ Happle, R.; Daniëls, O.; Koopman, R. J. J. (1993). "MIDAS syndrome (microphthalmia, dermal aplasia, and sclerocornea): an X-linked phenotype distinct from Goltz syndrome". ... "Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome" . Am. J. Hum. Genet . 79 (5): 878–89. doi : 10.1086/508474 . ... "Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome" . Am. J. Hum. Genet . 79 (5): 878–89. doi : 10.1086/508474 . ... External links [ edit ] Classification D OMIM : 309801 External resources GeneReviews : Microphthalmia with Linear Skin Defects Syndrome GeneReview/NIH/UW entry on Microphthalmia with Linear Skin Defects Syndrome This dermatology article is a stub .HCCS, NDUFB11, COX7B, ARHGAP6, FUS, DDIT3, CD6, GOLPH3, EWSR1, MXLPO, VPS13A, IL24, PRG4, STARD13, MIR135B, PORCN, THBS2, MIR486-1, LOC110806263, INTS1, SMUG1, YAP1, XK, WNT1, TP53, THPO, BIRC5, SOAT1, MET, CASP9, CDK4, CLCN4, ERG, GLI1, HSP90AA1, HTC2, MMP2, RET, SERPINE1, PIK3CA, PIK3CB, PIK3CD, PIK3CG, ARHGAP1, RASA1, PPARG
-
Myeloproliferative Syndrome, Transient
Omim
Transient myeloproliferative syndrome is a leukemoid reaction that occurs in some newborn infants with Down syndrome and rarely in phenotypically normal infants (Seibel et al., 1984). In 9 Down syndrome patients with transient myeloproliferative syndrome, Niikawa et al. (1991) found that the mode of inheritance of centromeric chromosomal markers was compatible with duplication of one parental chromosome 21. ... Cytogenetic and molecular studies demonstrated that in Down syndrome associated with transient abnormal myelopoiesis, trisomy 21 had arisen much more frequently through mitotic (or meiosis II) nondisjunctions than through meiosis I errors (Shen et al., 1995). ... The existence of a fusion gene is unlikely because almost no cases of TAM in Down syndrome had been reported with a rearranged chromosome 21 such as was observed in this critical case. Inheritance - Possible disomic homozygosity at 21q11.2 Misc - Usually in newborns with Down syndrome - rarely in normals Lab - Leukocytosis Heme - Transient myeloproliferative syndrome ▲ CloseGATA1, TTN, TAM, GAS6, ESR1, CAPN3, CCL2, TGFB1, MSTO1, CTNNB1, NFKB1, MPST, VEGFA, MAP3K10, KISS1R, WT1, AURKB, CCNE2, SQSTM1, ARHGEF7, CMYA5, NRIP1, YY1, VCP, VIM, SEMA3A, TYRO3, KANSL1, TP53, TNF, THPO, MIR100, SH2B3, MERTK, NDC80, NUP62, ORAI1, QRSL1, GOLM1, DCTN4, ASCC1, DLL1, DESI1, INTU, BBC3, UCN3, MAP1LC3B, PRAME, SIRT3, SIRT4, CTCF, SLC35G1, ZEB1, KHDRBS1, LAMTOR5, CLEC10A, ABL1, PCNA, CCL5, CDK1, FANCD2, EZH2, ERG, EGFR, CYP19A1, CYP2D6, CUX1, COMT, COL4A3, CD40, NR5A1, CASP3, FOXL2, BCL2, CCND1, AXL, ARHGDIA, APP, APOE, APC, FOXM1, GHR, PTPN11, JAK3, PSMB6, AKT1, OVGP1, MYCN, MPL, MMP9, MDM2, KIT, KISS1, ITGAL, GPER1, ITGA2B, CXCR2, CXCL8, IL6, IL3, IGFBP1, IGF1, HSPA5, GTF2H1, H3P40
-
White Sutton Syndrome
Wikipedia
Not to be confused with Sutton's disease . White–Sutton syndrome (WHSUS) is a rare neurodevelopmental disorder that affects different systems of the human body. ... In June 2016, the Online Mendelian Inheritance in Man (OMIM) designated this as “White–Sutton syndrome”. References [ edit ] ^ a b Reference, Genetics Home. "White-Sutton syndrome" . Genetics Home Reference . Retrieved 2018-11-23 . ^ "WHITE-SUTTON SYNDROME; WHSUS | MENDELIAN.CO" . www.mendelian.co (in Spanish) . Retrieved 2018-11-23 . ^ "OMIM Entry - # 616364 - WHITE-SUTTON SYNDROME; WHSUS" . www.omim.org . Retrieved 2018-11-23 . ^ Assia Batzir N, Posey JE, Song X, Akdemir ZC, Rosenfeld JA, Brown CW, Chen E, Holtrop SG, Mizerik E, Nieto Moreno M, Payne K, Raas-Rothschild A, Scott R, Vernon HJ, Zadeh N, Lupski JR, Sutton VR (2019) Phenotypic expansion of POGZ-related intellectual disability syndrome (White-Sutton syndrome). ... "POGZ truncating alleles cause syndromic intellectual disability" . Genome Medicine . 8 (1): 3. doi : 10.1186/s13073-015-0253-0 .
-
Knobloch Syndrome
Medlineplus
Knobloch syndrome is a rare condition characterized by severe vision problems and a skull defect. A characteristic feature of Knobloch syndrome is extreme nearsightedness (high myopia ). ... Due to abnormalities in the vitreous, retina, and macula, people with Knobloch syndrome often develop blindness in one or both eyes. ... In other conditions, encephaloceles may be associated with intellectual disability; however, most people with Knobloch syndrome have normal intelligence. Frequency Knobloch syndrome is a rare condition. ... Although the process is unclear, the COL18A1 gene mutations result in the loss of collagen XVIII protein, which likely causes the signs and symptoms of Knobloch syndrome. When the condition is caused by COL18A1 gene mutations, it is sometimes referred to as Knobloch syndrome type I.
-
Lateral Meningocele Syndrome
Wikipedia
Lateral meningocele syndrome Other names Lehman syndrome [1] Lateral meningocele syndrome is inherited in an autosomal dominant manner The lateral meningocele syndrome is a very rare skeletal disorder with facial anomalies, hypotonia and meningocele-related neurologic dysfunction. [2] Contents 1 Presentation 2 Genetics 3 Diagnosis 4 Treatment 5 History 6 References 7 External links Presentation [ edit ] Facial features found in this syndrome include dolichocephaly hypertelorism ptosis microretrognathia high arched palate long flat philtrum low set ears Non facial features of this syndrome include hyperextensibility hypotonia lateral meningoceles The lateral meningoceles are a common finding in this syndrome. They may be associated with neurological abnormalities and result in bladder dysfunction and neuropathy . Genetics [ edit ] This syndrome appears to be inherited in an autosomal dominant fashion. ... Mutations in Notch 3 were found to be associated with this syndrome. [3] Diagnosis [ edit ] This section is empty. ... References [ edit ] ^ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Lateral meningocele syndrome" . www.orpha.net . Retrieved 20 October 2019 . ^ Pamir, M. ... "Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome" . American Journal of Medical Genetics Part A . 167A (2): 271–81. doi : 10.1002/ajmg.a.36863 .NOTCH3, CDKN2A, TP53, ESR1, PSMB9, AKT1, ACTB, CALD1, PTDSS1, MYOCD, H3P10, IFNG, JAK1, KIT, MYC, MYLK, PIK3CD, PGR, PIK3CA, PIK3CB, GAS6, PIK3CG, PTEN, TYRO3, SLMAP, SMUG1, MDM2, DES, CASQ2, ACTG2, VCAN, H4C8, HDAC9, TP63, H4C14, H4C13, H4C5, MIR320A, H4C2, CD274, MVP, H4C3, H4C11, H4C12, H4C15, H4C6, H3P8, H4C4, H4C1, MIR152, MED12, MIRLET7D, SUB1, SF3B6, ANO1, HHAT, MIB1, PEG10, RPP14, CKAP4, H4C9, CARD14, LAMTOR2, SPINT2, TUBB3, AZIN2, H4-16, GADL1, OSTN, LINC01194, CDK2AP2, FZD6, ABL1, NAA10, LDHA, HTC2, HRAS, FABP3, EZH2, ELN, DMD, TIMM8A, DAP, CTNNB1, CSF3, CSF1, COMT, CFL2, CDSN, CD34, CCND2, BCL6, AXL, ALDH1A1, KRAS, MAA, TAM, MKI67, MLRL, UVRAG, TP53BP1, TOP2A, STAT3, STAT1, SRF, SRC, SLC12A3, SMTN, S100A9, RPE65, RB1, PTK2, PSME1, MAP2K7, PLAGL1, PCNA, ROR2, SPINT1

-
Achondrogenesis Type 2
Wikipedia
National Library of Medicine External links [ edit ] Classification D ICD - 10 : Q77.0 OMIM : 200610 DiseasesDB : 32635 v t e Osteochondrodysplasia Osteodysplasia/ / osteodystrophy Diaphysis Camurati–Engelmann disease Metaphysis Metaphyseal dysplasia Jansen's metaphyseal chondrodysplasia Schmid metaphyseal chondrodysplasia Epiphysis Spondyloepiphyseal dysplasia congenita Multiple epiphyseal dysplasia Otospondylomegaepiphyseal dysplasia Osteosclerosis Raine syndrome Osteopoikilosis Osteopetrosis Other/ungrouped FLNB Boomerang dysplasia Opsismodysplasia Polyostotic fibrous dysplasia McCune–Albright syndrome Chondrodysplasia / chondrodystrophy (including dwarfism ) Osteochondroma osteochondromatosis Hereditary multiple exostoses Chondroma / enchondroma enchondromatosis Ollier disease Maffucci syndrome Growth factor receptor FGFR2 : Antley–Bixler syndrome FGFR3 : Achondroplasia Hypochondroplasia Thanatophoric dysplasia COL2A1 collagen disease Achondrogenesis type 2 Hypochondrogenesis SLC26A2 sulfation defect Achondrogenesis type 1B Autosomal recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia Chondrodysplasia punctata Rhizomelic chondrodysplasia punctata Conradi–Hünermann syndrome Other dwarfism Fibrochondrogenesis Short rib – polydactyly syndrome Majewski's polydactyly syndrome Léri–Weill dyschondrosteosis v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteins
-
Cramp Fasciculation Syndrome
Wikipedia
Cramp fasciculation syndrome (CFS) is a rare [1] peripheral nerve hyperexcitability disorder. ... "Estimation of the frequency of the muscular pain-fasciculation syndrome and the muscular cramp-fasciculation syndrome in the adult population". ... "Cramp-fasciculation syndrome: a treatable hyperexcitable peripheral nerve disorder". ... "Bronchial involvement in the cramp-fasciculation syndrome". European Neurology . 56 (2): 124–6. doi : 10.1159/000095703 . ... D. (1991). "Cramp-fasciculation syndrome: A treatable hyperexcitable peripheral nerve disorder".
-
L1 Syndrome
Medlineplus
L1 syndrome describes a group of conditions that primarily affect the nervous system and occur almost exclusively in males. ... Individuals with MASA syndrome may have mild enlargement of the ventricles. ... Family members with L1 syndrome caused by the same mutation may have different forms of the condition. ... Causes L1 syndrome is caused by mutations in the L1CAM gene. ... These mutations typically lead to less severe cases of L1 syndrome, usually MASA syndrome or the other milder forms of this condition.
-
Prader-Willi Syndrome
Medlineplus
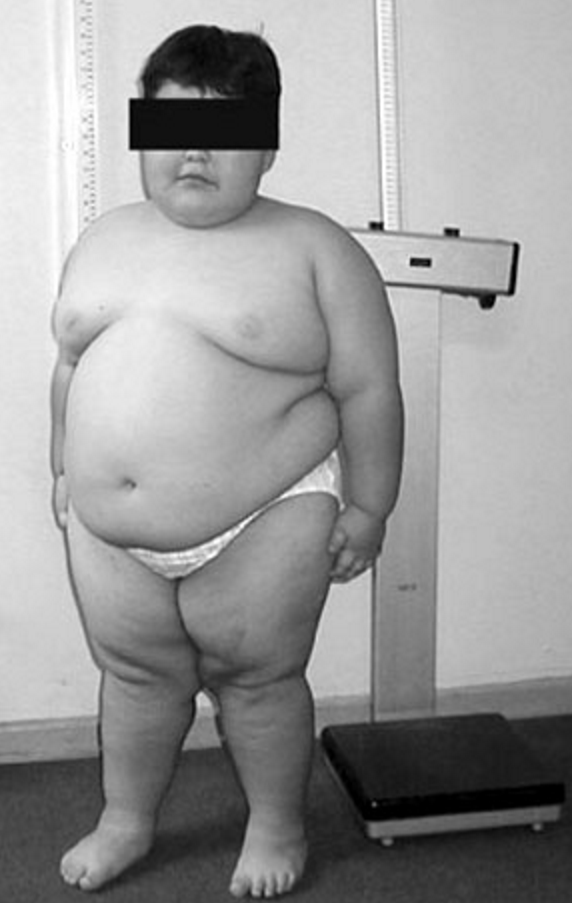
Prader-Willi syndrome is a complex genetic condition that affects many parts of the body. ... People with Prader-Willi syndrome typically have mild to moderate intellectual impairment and learning disabilities. ... Causes Prader-Willi syndrome is caused by the loss of function of genes in a particular region of chromosome 15 . ... In some people with Prader-Willi syndrome, the loss of a gene called OCA2 is associated with unusually fair skin and light-colored hair . ... Learn more about the gene and chromosome associated with Prader-Willi syndrome OCA2 chromosome 15 Inheritance Pattern Most cases of Prader-Willi syndrome are not inherited, particularly those caused by a deletion in the paternal chromosome 15 or by maternal uniparental disomy .MAGEL2, SNRPN, NDN, MKRN3, NPAP1, IPW, PWAR1, SNORD116-1, MKRN3-AS1, PWRN1, SNORD115-1, MRAP2, HERC2, HTR2C, CEP63, CENPJ, SOS1, PTPN11, RIT1, KRAS, RAF1, LZTR1, ATR, GH1, UBE3A, GABRB3, SNORD116@, UROD, SNURF, LEP, SNORD14D, SNORD14B, SNORD35B, SNORD14E, SNORD14C, GHRL, SNORD15A, IGF1, HCRT, GHR, SLC52A2, OCA2, F2R, NR1I2, ADIPOQ, PCSK1, ATP10A, POMC, SETDB1, IL6, GHRH, SNHG14, MAGED1, PWAR5, DMD, BEST1, ZNF274, FMR1, GNAQ, GLP1R, EHMT1, STOML3, HSPG2, STS, RASA1, TNFSF11, CRP, SLC6A4, LEPR, MARK2, TNFRSF11B, EHMT2, METAP2, NHLH2, GHSR, GABRA5, SOST, SIM1, PIK3CA, DKK1, PROCR, PADI4, EID1, RNF13, CIT, ABCB6, GREM1, CYFIP1, ACADS, ADIPOR1, RCBTB1, ZGLP1, MIR23A, MIR122, GOLGA8EP, RBMY1D, ENHO, PWARSN, RBMY2DP, GOLGA6A, WHAMMP3, SNORD109B, SNORD109A, NIPA1, TUBGCP5, LMLN, TMPRSS13, NIPA2, DHDDS, POM121, ADIPOR2, ANKRD36B, CHPT1, PRDM9, RETN, NSMCE3, ANGPTL8, ZNF654, DHX40, SMN2, PREPL, MSMB, MECP2, STMN1, HTR2B, HTC2, NR4A1, HDC, GCG, GAPDH, GABRG3, FRAXA, FMO2, EPHB1, EFNB2, DIO3, DAZ1, DAG1, CNR1, CD36, BTF3P11, BRAF, BGLAP, BDNF, BCR, AVP, ARSD, ARSA, AR, APOC3, ALCAM, MEN1, MST1, DLK1, CYTB, SCG2, VEGFA, TYR, TWIST1, TSPY1, TNF, THAS, STATH, STAR, AGRP, SMN1, SLC5A2, CCL5, SAG, RBMY1A1, PYY, PRNP, PRL, PRKCA, PTPA, PML, PIK3CG, PIK3CD, PIK3CB, PGC, PDPK1, NPY2R, NPY, HNRNPM, FMR1-IT1
-
Tetra-Amelia Syndrome
Wikipedia
Unsourced material may be challenged and removed. Find sources: "Tetra-amelia syndrome" – news · newspapers · books · scholar · JSTOR ( January 2019 ) ( Learn how and when to remove this template message ) Tetra-amelia syndrome Other names Autosomal recessive tetraamelia Violetta , a performer from the 1920s with tetra-amelia syndrome Tetra-amelia syndrome ( tetra- + amelia ), also called autosomal recessive tetraamelia , [1] is an extremely rare autosomal recessive [2] congenital disorder characterized by the absence of all four limbs. ... Because children with tetra-amelia syndrome have such serious medical problems, most are stillborn or die shortly after birth. Cause [ edit ] Tetra-amelia syndrome has an autosomal recessive pattern of inheritance . ... Mutations in the WNT3 gene prevent cells from producing functional WNT3 protein, which disrupts normal limb formation and leads to the other serious birth defects associated with tetra-amelia syndrome. Genetics within families [ edit ] In some affected families, the cause of tetra-amelia syndrome has not been determined. ... Epidemiology [ edit ] Tetra-amelia syndrome has been reported in only a few families worldwide.

-
Myhre Syndrome
Medlineplus
Myhre syndrome is a rare condition that affects connective tissue . ... Growth is reduced in most people with Myhre syndrome, beginning before birth and continuing through adolescence. ... Hearing loss occurs in most people with Myhre syndrome, usually beginning in childhood and gradually worsening. ... Frequency Myhre syndrome is a rare disorder; its prevalence is unknown. ... Increased SMAD4 availability allows the protein more time to interact with other proteins and may result in abnormal TGF-β signaling in many cell types, which affects development of several body systems and leads to the signs and symptoms of Myhre syndrome. Learn more about the gene associated with Myhre syndrome SMAD4 Inheritance Pattern Myhre syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered SMAD4 gene in each cell is sufficient to cause the disorder.
-
Löffler's Syndrome
Wikipedia
Löffler's syndrome Other names Loeffler's syndrome Eosinophil Specialty Respirology Löffler's syndrome is a disease in which eosinophils accumulate in the lung in response to a parasitic infection. ... Löffler's syndrome itself will cause difficulty breathing, coughing as well as a fever. ... Finally in 1943 the condition was called tropical eosinophilia by RJ Weingarten, and later officially named Löffler's syndrome. [9] The most well-known case of Löffler's syndrome was in a young boy from Louisiana. ... Retrieved 2018-12-05 . ^ "Löffler's syndrome" . British Medical Journal . 3 (5618): 569–570. 1968-09-07. ... Retrieved 2018-12-05 . ^ a b "Löffler's syndrome" . British Medical Journal . 3 (5618): 569–570. 1968-09-07.

-
Flammer Syndrome
Wikipedia
Syndrome of vascular dysregulation Flammer syndrome Flammer syndrome is a recently described clinical entity comprising a complex of clinical features caused mainly by dysregulation of the blood supply which has previously been called vascular dysregulation. ... People with Flammer syndrome typically respond excessively to mental or physical stimuli such as stress or exposure to cold. ... The blood vessels of individuals suffering from vasospastic syndrome respond to stimuli insufficiently. ... "The primary vascular dysregulation syndrome: implications for eye diseases" . ... External links [ edit ] Website on Flammer syndrome Scientific essay on Flammer syndrome
-
Abdominal Compartment Syndrome
Wikipedia
Abdominal compartment syndrome Specialty Emergency medicine Abdominal compartment syndrome ( ACS ) occurs when the abdomen becomes subject to increased pressure reaching past the point of intra-abdominal hypertension (IAH). ... There is a high mortality rate associated with abdominal compartment syndrome. [4] [10] Diagnosis [ edit ] Abdominal compartment syndrome is defined as an intra-abdominal pressure above 20 mmHg with evidence of organ failure. ... "Understanding abdominal compartment syndrome". Intensive Care Medicine . 42 (6): 1068–1070. doi : 10.1007/s00134-015-4089-2 . ... "Abdominal Compartment Syndrome". Surgery Today . 38 (1): 5–19. doi : 10.1007/s00595-007-3573-x . ... "Understanding abdominal compartment syndrome". Intensive Care Medicine . 42 (6): 1068–1070. doi : 10.1007/s00134-015-4089-2 .
-
Felty's Syndrome
Wikipedia
Autoimmune disease Felty's syndrome Specialty Rheumatology Felty's syndrome , also called Felty syndrome , ( FS ) [1] is rare autoimmune disease characterized by the triad of rheumatoid arthritis , enlargement of the spleen and too few neutrophils in the blood . ... In some affected individuals, Felty's syndrome may develop during a period when the symptoms and physical findings associated with rheumatoid arthritis have subsided or are not present; in this case, Felty's syndrome may remain undiagnosed. ... Keratoconjunctivitis sicca may occur due to secondary Sjögren's syndrome . Individuals with Felty's syndrome may also experience fever, weight loss, and/or fatigue. ... People with this syndrome are at risk of infection because they have a low white blood cell count. [5] Mechanism [ edit ] The underlying pathogenesis of Felty's syndrome is not clear. ... PMID 15454123 . ^ a b Woolston, W 2017, 'Felty's Syndrome: A Qualitative Case Study', MEDSURG Nursing , vol. 26, no. 2, pp. 105-118. ^ "CIGNA – Felty's Syndrome" .
-
Dup15q
Wikipedia
Dup15q syndrome is the common name for chromosome 15q11.2-q13.1 duplication syndrome. ... Important genes likely involved in the etiology of Dup15q syndrome include UBE3A , GABRA5 , GABRB3 , and GABRG3 . ... The global chromosome 15q11.2-13.1 duplication syndrome specific groups only provide medical information and research for chromosome 15q11.2-13.1 duplication syndrome and not the outlying 15q duplications. ... Within the Dup15q syndrome cohort, children with epilepsy had greater cognitive impairment. ... Two duplication types are commonly described in Dup15q syndrome, interstitial and isodicentric.
-
Hypermobility Spectrum Disorder
Wikipedia
Hypermobility spectrum disorder Other names Hypermobility syndrome joint hypermobility syndrome Specialty Rheumatology , genetics Symptoms Joint hypermobility , musculoskeletal pain , fatigue [1] Causes Genetic [2] Hypermobility spectrum disorder ( HSD ), related to earlier diagnoses such as hypermobility syndrome ( HMS ), and joint hypermobility syndrome ( JHS ) is a heritable connective tissue disorder [3] that affects joints and ligaments. ... In particular, musculoskeletal involvement is a requirement for diagnosis with any form of hypermobility spectrum disorder but not for hypermobile Ehlers-Danlos syndrome. Like hypermobile Ehlers-Danlos syndrome, hypermobility spectrum disorders are associated with orthostatic tachycardia , gastrointestinal disorders, and pelvic and bladder dysfunction. [11] Treatment [ edit ] Treating hypermobility syndrome can be difficult. ... "Hypermobility Disorders- An Update for Clinicans" . Hypermobility Syndromes Association . Retrieved 30 May 2018 . ^ "Hypermobile Ehlers-Danlos syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ... Tinkle, Issues and Management of Joint Hypermobility. A Guide for the Ehlers-danlos Syndrome Hypermobility Type and the Hypermobility Syndrome , Left Paw Press, USA (2008) ^ a b Palmer et al., 2017 ^ a b c Castori, Marco; Tinkle, Brad; Levy, Howard; Grahame, Rodney; Malfait, Fransiska; Hakim, Alan (March 2017). ... "A study exploring the prevalence of joint hypermobility syndrome in patients attending a musculoskeletal triage clinic."
-
Osteopoikilosis
Wikipedia
Additionally, the disease is often associated with melorheostosis , [4] despite the apparent lack of correlation between melorheostosis and genetic heritability. [ citation needed ] It has been tied to LEMD3 . [5] Buschke–Ollendorff syndrome is a similar condition, [6] which is also associated with LEMD3 . [7] Osteopoikilosis of the hips on CT. ... "Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke–Ollendorff syndrome and melorheostosis" . Nat. Genet . 36 (11): 1213–8. doi : 10.1038/ng1453 . ... "Deactivating germline mutations in LEMD3 cause osteopoikilosis and Buschke–Ollendorff syndrome, but not sporadic melorheostosis". ... External links [ edit ] Classification D ICD - 10 : Q78.8 ICD - 9-CM : 756.53 OMIM : 166700 MeSH : D010023 DiseasesDB : 30071 External resources eMedicine : derm/733 v t e Osteochondrodysplasia Osteodysplasia/ / osteodystrophy Diaphysis Camurati–Engelmann disease Metaphysis Metaphyseal dysplasia Jansen's metaphyseal chondrodysplasia Schmid metaphyseal chondrodysplasia Epiphysis Spondyloepiphyseal dysplasia congenita Multiple epiphyseal dysplasia Otospondylomegaepiphyseal dysplasia Osteosclerosis Raine syndrome Osteopoikilosis Osteopetrosis Other/ungrouped FLNB Boomerang dysplasia Opsismodysplasia Polyostotic fibrous dysplasia McCune–Albright syndrome Chondrodysplasia / chondrodystrophy (including dwarfism ) Osteochondroma osteochondromatosis Hereditary multiple exostoses Chondroma / enchondroma enchondromatosis Ollier disease Maffucci syndrome Growth factor receptor FGFR2 : Antley–Bixler syndrome FGFR3 : Achondroplasia Hypochondroplasia Thanatophoric dysplasia COL2A1 collagen disease Achondrogenesis type 2 Hypochondrogenesis SLC26A2 sulfation defect Achondrogenesis type 1B Autosomal recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia Chondrodysplasia punctata Rhizomelic chondrodysplasia punctata Conradi–Hünermann syndrome Other dwarfism Fibrochondrogenesis Short rib – polydactyly syndrome Majewski's polydactyly syndrome Léri–Weill dyschondrosteosis v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins
-
Pulmonary-Renal Syndrome
Wikipedia
Pulmonary-renal syndrome (PRS) is a rare medical syndrome in which respiratory failure involving bleeding in the lungs and kidney failure ( glomerulonephritis ) occur. [1] PRS is associated with a high rate of morbidity and death. [1] The term was first used by Goodpasture in 1919 to describe the association of respiratory and kidney failure. [1] Contents 1 Causes 2 Diagnosis 2.1 Differential diagnosis 3 Treatment 4 References Causes [ edit ] Pulmonary-renal syndromes are most commonly caused by an underlying autoimmune disease. PRS is most commonly due to ANCA -associated vasculitides (e.g., granulomatosis with polyangiitis ) or due to anti-basement membrane diseases (e.g., Goodpasture's syndrome ). Granulomatosis with polyangiitis usually presents with nasopharyngeal involvement as well, whereas Goodpasture's will not. ... Other etiologies include toxic injury such as paraquat poisoning, infection with hantavirus , leptospirosis , or legionella , or vascular, as seen in nephrotic syndrome when a renal vein thrombosis embolizes to the lungs. ... Plasmapheresis can be used in some circumstances. [ citation needed ] References [ edit ] ^ a b c d e West, SC; Arulkumaran, N; Ind, PW; Pusey, CD (May 2013). "Pulmonary-renal syndrome: a life threatening but treatable condition". ... PMID 23349383 . v t e Diseases of the respiratory system Upper RT (including URTIs , common cold ) Head sinuses Sinusitis nose Rhinitis Vasomotor rhinitis Atrophic rhinitis Hay fever Nasal polyp Rhinorrhea nasal septum Nasal septum deviation Nasal septum perforation Nasal septal hematoma tonsil Tonsillitis Adenoid hypertrophy Peritonsillar abscess Neck pharynx Pharyngitis Strep throat Laryngopharyngeal reflux (LPR) Retropharyngeal abscess larynx Croup Laryngomalacia Laryngeal cyst Laryngitis Laryngopharyngeal reflux (LPR) Laryngospasm vocal cords Laryngopharyngeal reflux (LPR) Vocal fold nodule Vocal fold paresis Vocal cord dysfunction epiglottis Epiglottitis trachea Tracheitis Laryngotracheal stenosis Lower RT / lung disease (including LRTIs ) Bronchial / obstructive acute Acute bronchitis chronic COPD Chronic bronchitis Acute exacerbation of COPD ) Asthma ( Status asthmaticus Aspirin-induced Exercise-induced Bronchiectasis Cystic fibrosis unspecified Bronchitis Bronchiolitis Bronchiolitis obliterans Diffuse panbronchiolitis Interstitial / restrictive ( fibrosis ) External agents/ occupational lung disease Pneumoconiosis Aluminosis Asbestosis Baritosis Bauxite fibrosis Berylliosis Caplan's syndrome Chalicosis Coalworker's pneumoconiosis Siderosis Silicosis Talcosis Byssinosis Hypersensitivity pneumonitis Bagassosis Bird fancier's lung Farmer's lung Lycoperdonosis Other ARDS Combined pulmonary fibrosis and emphysema Pulmonary edema Löffler's syndrome / Eosinophilic pneumonia Respiratory hypersensitivity Allergic bronchopulmonary aspergillosis Hamman-Rich syndrome Idiopathic pulmonary fibrosis Sarcoidosis Vaping-associated pulmonary injury Obstructive / Restrictive Pneumonia / pneumonitis By pathogen Viral Bacterial Pneumococcal Klebsiella Atypical bacterial Mycoplasma Legionnaires' disease Chlamydiae Fungal Pneumocystis Parasitic noninfectious Chemical / Mendelson's syndrome Aspiration / Lipid By vector/route Community-acquired Healthcare-associated Hospital-acquired By distribution Broncho- Lobar IIP UIP DIP BOOP-COP NSIP RB Other Atelectasis circulatory Pulmonary hypertension Pulmonary embolism Lung abscess Pleural cavity / mediastinum Pleural disease Pleuritis/pleurisy Pneumothorax / Hemopneumothorax Pleural effusion Hemothorax Hydrothorax Chylothorax Empyema/pyothorax Malignant Fibrothorax Mediastinal disease Mediastinitis Mediastinal emphysema Other/general Respiratory failure Influenza Common cold SARS Coronavirus disease 2019 Idiopathic pulmonary haemosiderosis Pulmonary alveolar proteinosis v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy v t e Hypersensitivity and autoimmune diseases Type I / allergy / atopy ( IgE ) Foreign Atopic eczema Allergic urticaria Allergic rhinitis (Hay fever) Allergic asthma Anaphylaxis Food allergy common allergies include: Milk Egg Peanut Tree nut Seafood Soy Wheat Penicillin allergy Autoimmune Eosinophilic esophagitis Type II / ADCC IgM IgG Foreign Hemolytic disease of the newborn Autoimmune Cytotoxic Autoimmune hemolytic anemia Immune thrombocytopenic purpura Bullous pemphigoid Pemphigus vulgaris Rheumatic fever Goodpasture syndrome Guillain–Barré syndrome " Type V "/ receptor Graves' disease Myasthenia gravis Pernicious anemia Type III ( Immune complex ) Foreign Henoch–Schönlein purpura Hypersensitivity vasculitis Reactive arthritis Farmer's lung Post-streptococcal glomerulonephritis Serum sickness Arthus reaction Autoimmune Systemic lupus erythematosus Subacute bacterial endocarditis Rheumatoid arthritis Type IV / cell-mediated ( T cells ) Foreign Allergic contact dermatitis Mantoux test Autoimmune Diabetes mellitus type 1 Hashimoto's thyroiditis Multiple sclerosis Coeliac disease Giant-cell arteritis Postorgasmic illness syndrome Reactive arthritis GVHD Transfusion-associated graft versus host disease Unknown/ multiple Foreign Hypersensitivity pneumonitis Allergic bronchopulmonary aspergillosis Transplant rejection Latex allergy (I+IV) Autoimmune Sjögren syndrome Autoimmune hepatitis Autoimmune polyendocrine syndrome APS1 APS2 Autoimmune adrenalitis Systemic autoimmune disease This article about a medical condition affecting the respiratory system is a stub .HLA-DRB1, RBM45, COL4A3, HLA-DQB1, MPO, FCGR2B, FCGR3A, FCGR3B, FCGR2A, DDR1, IGAN1, B3GAT1, CERT1, PXDN, TNF, TGFB1, SYK, RPS19, PPARA, ABO, MMP12, LAMA5, IL1RN, BTK, HLA-DQB2, HLA-DPB1, HLA-C, CFH, GNAO1, EGFR, ATN1, WG






