See also ovarian dysgenesis with sensorineural deafness, or Perrault syndrome (233400). Clinical Features Elliott et al. (1959) reported the condition in 3 sisters who had normal stature and sex chromatin but had never menstruated and had severe osteoporosis. ... Each had a normal female 46,XX karyotype. Somatic features of Turner syndrome were not found. All 3 had elevated gonadotropins, and laparotomy on the 2 older sisters showed streak gonads and unstimulated mullerian structures.
A number sign (#) is used with this entry because of evidence that ovarian dysgenesis-7 (ODG7) is caused by homozygous mutation in the MRPS22 gene (605810) on chromosome 3q23. Description Ovarian dysgenesis-7 is characterized by primary amenorrhea, delayed puberty, elevated gonadotropic hormones, and small uterus and ovaries. Ovarian histology shows fibrotic ovaries without follicles (Chen et al., 2018). For a discussion of genetic heterogeneity of ovarian dysgenesis, see ODG1 (233300). Clinical Features Chen et al. (2018) described 2 46,XX sisters and their female cousin from a large consanguineous Israeli-Christian Arab family, who had delayed puberty with elevated gonadotropins and small uterus and ovaries.
A number sign (#) is used with this entry because this form of hypergonadotropic ovarian dysgenesis (ODG3) is caused by homozygous mutation in the PSMC3IP gene (608665) on chromosome 17q12-q21. For a discussion of genetic heterogeneity of hypergonadotropic ovarian dysgenesis, see ODG1 (233300). Clinical Features Zangen et al. (2011) studied a large consanguineous Arab Palestinian pedigree in which at least 5 females were affected with complete XX gonadal dysgenesis. The proband failed to develop spontaneous puberty and at 15 years of age, breast development and pubic hair were at Tanner stage 1 and 2, respectively; hormonal testing revealed high basal gonadotropin levels with undetectable estradiol and progesterone and normal levels of androgens. Her karyotype was 46,XX with no SRY sequence detected, and abdominal ultrasound and MRI showed uterine hypoplasia and undetectable ovaries.
A number sign (#) is used with this entry because of evidence that ovarian dysgenesis-6 (ODG6) is caused by homozygous mutation in the NUP107 gene (607617) on chromosome 12q15. One such family has been reported. Description Ovarian dysgenesis-6 is characterized by absence of spontaneous pubertal development in females with elevated gonadotropin levels, small uterus, and absence of ovarian tissue on imaging studies. Males appear to be unaffected (Weinberg-Shukron et al., 2015). For a discussion of genetic heterogeneity of ovarian dysgenesis, see ODG1 (233300). Clinical Features Weinberg-Shukron et al. (2015) studied a girl from a large consanguineous Palestinian family who presented at age 15 years with absence of spontaneous puberty and had minimal breast development, pubertal hair at Tanner stage II, primary amenorrhea, and elevated luteinizing hormone (LH; see 152780) and follicle-stimulating hormone (FSH; see 136530) levels. Ultrasound and MRI of the pelvis showed a relatively small uterus, and ovaries were not detected.
A number sign (#) is used with this entry because of evidence that ovarian dysgenesis-2 (ODG2) is caused by mutation in the BMP15 gene (300247) on chromosome Xp11. Mutation in the BMP15 gene can also cause premature ovarian failure-4 (POF4). For a discussion of genetic heterogeneity of ovarian dysgenesis, see ODG1 (233300). For a phenotypic description and a discussion of genetic heterogeneity of premature ovarian failure, see POF1 (311360). Description Hypergonadotropic ovarian failure is a heterogeneous disorder that, in the most severe forms, is a result of ovarian dysgenesis.
Description CDGs, previously called carbohydrate-deficient glycoprotein syndromes, grew from hereditary multisystem disorders first recognized by Jaeken et al. (1980). ... The patient was referred at 4 months of age for edematoascitic syndrome related to severe hypoalbuminemia resulting from protein-losing enteropathy.
A form of congenital disorders of N-linked glycosylation that is characterized by gastrointestinal symptoms (diarrhea, vomiting, feeding problems with failure to thrive, protein-losing enteropathy), edema and ascites (including hydrops fetalis), hepatomegaly, renal tubulopathy, coagulation anomalies due to thrombocytopenia, brain involvement (psychomotor delay, seizures, ataxia), facial dysmorphism (low-set ears and retrognathia), pes equinovarus, and muscular hypotonia. Cataracts may also be observed. Prognosis is usually poor. The disease is caused by loss-of-function mutations in the gene ALG8 (11q14.1), resulting in a block in the initial step of protein glycosylation.
TBG defects are clinically fully expressed in hemizygous males and rarely in Turner syndrome or homozygous females, whereas in heterozygous females TBG deficiency is usually partial. ... X-linked inheritance was strongly supported by the finding of deficiency of TBG in a patient with the XO Turner syndrome (Refetoff and Selenkow, 1968).
Tadiboyina et al. (2005) reported 3 patients from 2 apparently unrelated Old Colony Mennonite families, each of whom had the hepatocerebral form of mitochondrial DNA depletion syndrome (251800) together with cystathioninuria. ... Tadiboyina et al. (2005) suggested that the hepatocerebral form of mtDNA depletion syndrome might be associated with secondary cystathioninuria.
A rare inborn error of metabolism characterized by abnormal accumulation of plasma cystathionine and subsequent increased urinary excretion due to cystathionine gamma-lyase deficiency. The condition is considered benign without pathological relevance. Mode of inheritance is autosomal recessive.
One sister reported loss of pain and temperature sensation in the left arm, shoulder, and face since age 12 years. She also had right Horner syndrome, rotary nystagmus, and absent left gag reflex. ... INHERITANCE - Autosomal dominant HEAD & NECK Face - Facial pain - Facial numbness Eyes - Nystagmus - Horner syndrome Neck - Neck pain GENITOURINARY Bladder - Urinary incontinence SKELETAL Skull - Associated with abnormalities at the foramen magnum, especially Chiari malformation type I ( 118420 ) MUSCLE, SOFT TISSUES - Muscle atrophy in the limbs - Muscle weakness NEUROLOGIC Central Nervous System - Unsteady gait - Ataxic gait - Spasticity of the lower limbs - Hyperreflexia, especially of the lower limbs - Areflexia of the upper limbs - Upper limb weakness - Neck pain - Arm pain - Cranial nerve anomalies - Burning pain in the limbs - Loss of pain and temperature in a cape-like distribution - Touch, vibration, and limb position may or may not be affected - Segmental sensory loss, especially of pain and temperature - Paresthesias - Dysarthria - Extensor plantar responses - Chiari I malformation on MRI (some) MISCELLANEOUS - Onset of symptoms in second or third decade - Many cases are asymptomatic ▲ Close
Syringomyelia is a condition in which a cyst, called a syrinx, forms within the spinal cord. This cyst expands and elongates over time, destroying the center of the spinal cord which can result in pain, weakness, stiffness in the back, shoulders, arms, or legs, headaches, and insensitivity to temperature (especially in the hands). Symptoms vary from person to person. Syringomyelia is often related to a congenital abnormality of the brain called a Chiari I malformation, but may also occur as a complication of trauma, inflammation of the tissue that surrounds the brain and spinal cord ( meningitis) such as the inflammation of the arachnoides ( arachnoiditis ), hemorrhage , or a tumor. Symptoms may appear months or even years after the initial injury. Some cases of syringomyelia are familial, although this is rare. Treatment often involves surgery and avoiding activities that involve straining.
A significantly increased risk of parathyroid cancer is also a feature of certain rare genetic syndromes. Parathyroid cancer occurs in 15 percent of individuals with hyperparathyroidism-jaw tumor syndrome and in 1 percent of individuals with familial isolated hyperparathyroidism.
Molecular Genetics Shattuck et al. (2003) identified somatic and germline mutations in the CDC73 gene (607393) in patients who had no known family history of primary hyperparathyroidism (HPRT1, 145000) or the hyperparathyroidism-jaw tumor syndrome (HPRT2, HPT-JT; 145001), also caused by mutation in the CDC73 gene, at presentation.
A rare endocrine tumor characterized by a malignant neoplasm derived from parathyroid parenchymal cells, localized in one of the normally located parathyroid glands or other sites where parathyroid tissue may be present. Signs and symptoms are predominantly due to excess secretion of parathyroid hormone, with marked hypercalcemia and renal and bone involvement. In rare cases, the tumor may be non-functioning and only present as a palpable mass in the neck region. Recurrent laryngeal nerve paralysis is also observed. The tumor can occur sporadically or on a genetic background. The extent of invasion of adjacent structures positively correlates with the development of recurrent or metastatic disease.
A large mass in the neck is often seen, and kidney and bone abnormalities are common. [1] Risk factors [ edit ] Parathyroid cancer occurs in midlife at the same rate in men and women. [ citation needed ] Conditions that appear to result in an increased risk of parathyroid cancer include multiple endocrine neoplasia type 1 , [4] autosomal dominant familial isolated hyperparathyroidism [4] and hyperparathyroidism-jaw tumor syndrome [1] (which also is hereditary). [1] Parathyroid cancer has also been associated with external radiation exposure, but, most reports describe an association between radiation and the more common parathyroid adenoma . [4] Diagnosis [ edit ] This section is empty.
The authors noted that in the mouse, an obesity-diabetes syndrome elicited by a genetic defect in a prohormone processing pathway was described by Naggert et al. (1995); see 114855. Jackson et al. (1997) noted that 2 specific syndromes caused by abnormalities in the secretory products of pancreatic beta cells had been defined; mutations in the insulin gene that lead to the production of biologically ineffective insulin (see 176730.0005), and mutations in the insulin gene that affect the cleavage of proinsulin which lead to the secretion of excessive amounts of proinsulin (see 176730.0003).
A rare genetic endocrine disease characterized by early onset of severe intractable diarrhea and intestinal malabsorption, followed by obesity and hormonal deficiencies due to insufficient activation of several prohormones, resulting in hypocortisolism, hypothyroidism, diabetes insipidus, hypogonadism, growth deficiency, and diabetes mellitus. Extent and age of onset of hormone deficiencies are variable between patients.
Complex vertebral malformation or CVM is a lethal hereditary syndrome found in Holstein cattle . [1] [2] CVM is responsible for malformed calves that are either spontaneously aborted or die shortly after birth. [1] [3] It is caused by a missense mutation in the SLC35A3 gene. [2] Since the mutant form of the gene is recessive , only individuals carrying two copies of the faulty gene ( homozygous individuals) are affected. ... Carlin-M Ivanhoe Bell's grandsire Osborndale Ivanhoe, however, carried only BLAD. [2] Scientists therefore believe that the mutation responsible for CVM occurred either in Penstate Ivanhoe Starissa, or somewhere in his maternal family. [2] The CVM syndrome was first found in a Danish Holstein stock in 1999, and during the following years it was also found in the United States, United Kingdom, Netherlands, and Japan. [2] Because of the wide international usage of Carlin-M Ivanhoe Bell and the large number of animals descending from him, the CVM gene is found in Holstein cattle throughout the world.
ISBN 978-0-521-87593-6 . v t e Diseases of crustaceans Bitter crab disease Crayfish plague Gaffkaemia Infectious hypodermal and haematopoietic necrosis Necrotising hepatopancreatitis Paragonimiasis Taura syndrome White spot syndrome Yellowhead disease
There are also higher risks of carpal tunnel syndrome and osteoarthritis in patients with a previous Smith fractures. Entrapment of the extensor pollicus longus can also occur in cases of non-union, and can result in late rupture of this tendon. Complex regional pain syndrome can be reported in up to 40% of fractures. [4] Diagnosis [ edit ] Physical examination [ edit ] Classic physical examination findings of a Smith's fracture is palmar displacement of the wrist that results in a "garden-spade deformity".
This article is about the vertebral fracture. For the wrist fracture, see Smith's fracture . A Smith fracture is a named vertebral fracture occurring most commonly in the lumbar spine . It is similar to that of a Chance fracture and is associated with seat belt injuries. This fracture represents a fracture through the posterior elements including the superior articular processes but not the spinous process, as well as an avulsion fracture of the vertebral body. [1] It is not to be confused with the more commonly referred to Smith's fracture of the wrist. Notes [ edit ] ^ Richard H. Daffner - Chance-Type Fractures of the Thoracolumbar Spine: Imaging Analysis: Discussion References [ edit ] Smith NS, Kaufer H: Patterns and mechanisms of lumbar injuries associated with lapseat belts.
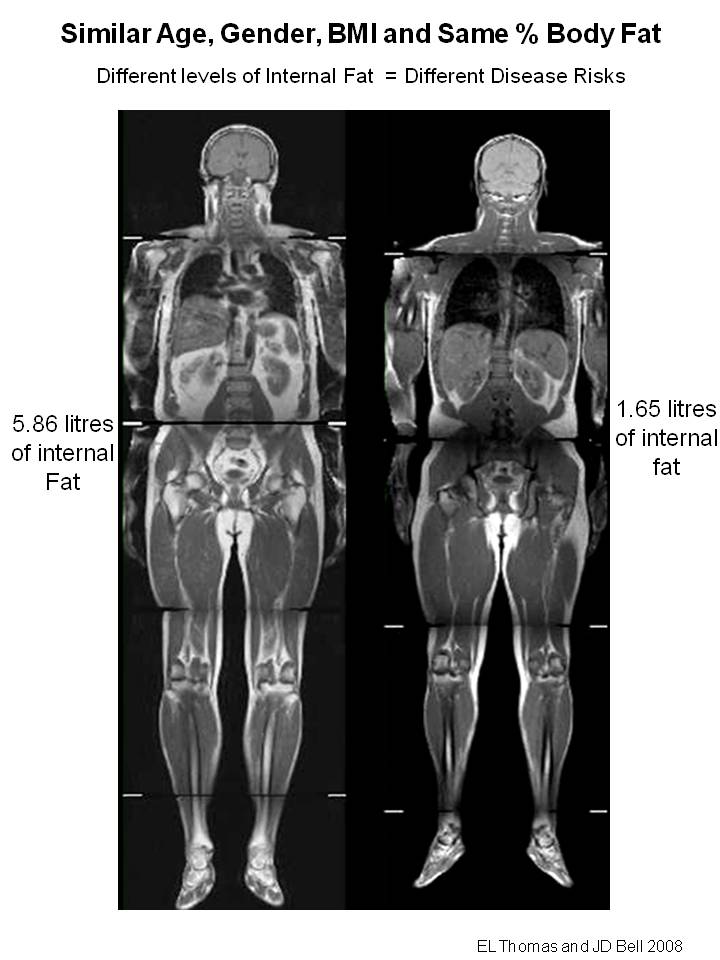
Subjects defined as TOFI with body mass index (BMI) <25 kg/m 2 have increased levels of many of the risk factors associated with the metabolic syndrome . This phenotype is a further refinement of “metabolically-obese normal-weight" [3] [4] [5] (MONW). ... "Normal weight obese (NWO) women: an evaluation of a candidate new syndrome". Nutrition, Metabolism & Cardiovascular Diseases . 16 (8): 513–523. doi : 10.1016/j.numecd.2005.10.010 .
Caseous lymphadenitis Other names Thin ewe syndrome Specialty Veterinary medicine Symptoms Pus-filled abscesses in lymph nodes and internal organs, weight loss Causes Corynebacterium pseudotuberculosis Treatment Drainage of abscesses, chemical cauterization, removal of external lymph nodes, antibiotics Caseous lymphadenitis is an infectious disease caused by the bacterium Corynebacterium pseudotuberculosis , that affects the lymphatic system , resulting in abscesses in the lymph nodes and internal organs . ... In only three species; sheep, goats and horses, it is recognized as a specific disease syndrome. The biotype of Corynebacterium pseudotuberculosis affecting horses and cattle is distinguishable from the biotype that infects small ruminants based on its ability to reduce nitrate in vitro .
Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( February 2017 ) Infantile apnea Specialty Pediatric Infantile apnea is a rare disease that is characterized by cessation of breathing in an infant for at least 20 seconds or a shorter respiratory pause that is associated with a slow heart rate , bluish discolouration of the skin , extreme paleness , gagging, choking and/or decreased muscle tone . [1] [2] Infantile apnea occurs in children under the age of one and it is more common in premature infants. [3] Symptoms of infantile apnea occur most frequently during the rapid eye movement (REM) stage of sleep. [4] The nature and severity of breathing problems in patients can be detected in a sleep study called a polysomnography which measures the brain waves, heartbeat, body movements and breathing of a patient overnight. [4] Infantile apnea can be caused by developmental problems that result in an immature brainstem or it can be caused other medical conditions. [1] [4] [5] As children grow and develop, infantile apnea usually does not persist. [4] Infantile apnea may be related to some cases of sudden infant death syndrome (SIDS) however, the relationship between infantile apnea and SIDS is not known. [3] Contents 1 Cause 2 Diagnosis 2.1 Classification 2.1.1 Central Apnea 2.1.2 Obstructive Apnea 2.1.3 Mixed Apnea 3 Epidemiology 4 References Cause [ edit ] √ having a family history of sleep apnea. √ being overweight or obese. ✓ having certain medical conditions (cerebral palsy, Down syndrome, sickle cell disease, abnormalities in the skull or face) ✓ being born with a low birth weight. ✓ having a large tongue.
Infantile apnea is a cessation of respiratory air flow that may affect newborns or older children because of neurological impairment of the respiratory rhythm or obstruction of air flow through the air passages. The symptoms include cyanosis, pallor or bradycardia and snoring in case of obstructive apnea.
Part of a series on Spirituality Outline Religion History Timeline Traditional Christian Catholic Mysticism Modern Buddhist modernism New religious movement Secular spirituality " Spiritual but not religious " Syncretism Spiritual experience Mystical experience Religious experience Spiritual practice Spiritual development Ego death Individuation Spiritual development Self-actualization Spiritual activism Influences Western General Divine illumination Pantheism Panentheism Antiquity Gnosticism Hermeticism Neoplatonism Western esotericism Medieval Mysticism Early modern Perennial philosophy Jakob Böhme Emanuel Swedenborg Pietism Modern Romanticism Transcendentalism Universalism New Thought Theosophy Anthroposophy Occultism Spiritualism Esoteric Christianity New Age Orientalist Comparative religion Neo-Advaita Nondualism Orientalism Theosophical Society Asian Pre-historic Proto-Indo-Iranian religion Iran Zoroastrianism India Advaita Vedanta Buddha-nature Enlightenment Madhyamaka Neo-Vedanta Tantra Yoga Yogachara East-Asia Taoism Other non-Western Animism Shamanism Totemism Psychological Humanistic psychology Mindfulness Positive psychology Self-help Self-realization True self and false self Research Neurological Mystical psychosis Cognitive science of religion Neuroscience of religion Geschwind syndrome Evolutionary psychology of religion Category v t e Mystical psychosis is a term coined by Arthur J. ... Deikman considered that all-encompassing unity opened in mysticism can be all-encompassing unity of reality. [15] See also [ edit ] Altered state of consciousness Depersonalization and Derealization Existential crisis Dhyāna in Buddhism Dhyāna in Hinduism Jerusalem syndrome Mental health Moksha Mirror neurons Mysticism Monomyth Near-death experience Posttraumatic stress disorder Religious experience Spiritualism Spirituality Spiritual crisis Wujud References [ edit ] ^ Whitney, E. (1998). " Personal accounts: Mania as spiritual emergency " Psychiatric Services 49 : 1547–1548 ^ Jackson, M., & Fulford, K.W.M., K.
Mucosal neuromas Cutaneous capillary blush Ipsilateral macroglossia Syndromic hemimegalencaphaly Not all patients with facial infiltrating lipomatosis have all these symptoms. ... Plastic and Reconstructive Surgery . 136 : 72–73. doi : 10.1097/01.prs.0000472371.96995.e5 . ^ Clinical trial number NCT03094832 for "Study of ARQ 092 in Subjects With PIK3CA-related Overgrowth Spectrum and Proteus Syndrome (MOSAIC)" at ClinicalTrials.gov ^ Clinical trial number NCT04085653 for "Managed Access Program (MAP) to Provide Alpelisib (BYL719) for Patients With PIK3CA-Related Overgrowth Spectrum (PROS)" at ClinicalTrials.gov
"Pedantic speaking style differentiates Asperger syndrome from high-functioning autism". ... "Speech and prosody characteristics of adolescents and adults with high-functioning autism and Asperger syndrome". Journal of Speech, Language, and Hearing Research . 44 (5): 1097–1115.
There were two factors that were considered: The specificity of the impairment: in SDD there is one single domain that is affected, whereas in PDD multiple areas of functioning are affected. [8] The nature of the impairment: development in SDD is delayed but not otherwise abnormal, whereas in PDD there are behavioral deviations that are not typical for any developmental stage. [8] In the DSM-IV, specific developmental disorders were no longer grouped together. [9] Instead they were reclassified as communication disorders, learning disorders, and motor skills disorders. [3] Comparison and conditions [ edit ] ICD-10 [10] DSM-IV-TR [11] ICD-11 [12] Specific developmental disorders of speech and language (F80): Specific speech articulation disorder (F80.0) Expressive language disorder (F80.1) Receptive language disorder (F80.2) Acquired aphasia with epilepsy Landau-Kleffner syndrome (F80.3) Other developmental disorders of speech and language (F80.8) Developmental disorder of speech and language, unspecified (F80.9) Communication disorders : Expressive Language Disorder (315.31) Mixed Receptive-Expressive Language Disorder (315.32) Phonological Disorder (315.39) Stuttering (307.0) Communication Disorder Not Otherwise Specified (307.9) Developmental Speech & Language Disorders (6A01): Developmental speech sound disorder (6A01.0) Developmental speech fluency disorder (6A01.1) Developmental language disorder (6A01.2) Developmental language disorder with impairment of receptive and expressive language (6A01.20) Developmental language disorder with impairment of mainly expressive language (6A01.21) Developmental language disorder with impairment of mainly pragmatic language (6A01.22) Developmental language disorder, with other specified language impairment (6A01.23) Other specified developmental speech or language disorders (6A01.Y) Developmental speech or language disorders, unspecified (6A01.Z) Specific developmental disorders of scholastic skills (F81): Specific reading disorder (F81.0) Specific spelling disorder (F81.1) Specific disorder of arithmetical skills (F81.2) Mixed disorder of scholastic skills (F81.3) Other disorders of scholastic skills (F81.8) Developmental disorder of scholastic skills, unspecified (F81.9) Learning disorders : Reading Disorder (315.0) Mathematics Disorder (315.1) Disorder of Written Expression (315.2) Learning Disorder Not Otherwise Specified (315.9) Developmental learning disorder (6A03): Developmental learning disorder with impairment in reading (6A03.0) Developmental learning disorder with impairment in written expression (6A03.1) Developmental learning disorder with impairment in mathematics (6A03.2) Developmental learning disorder with other specified impairment of learning (6A03.3) Developmental learning disorder, unspecified (6A03.Z) Specific developmental disorder of motor function (F82) Motor skills disorders : Developmental coordination disorder (315.4) Developmental motor coordination disorder (6A04) Mixed specific developmental disorder (F83) See also [ edit ] Developmental disability References [ edit ] ^ Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition . ... Taylor: Child and Adolescent Psychiatry , 4th ed. 2005 ^ Robert Jean Campbell, III: Campbell's Psychiatric Dictionary , 2003, page 184 ^ http://apps.who.int/classifications/icd10/browse/2010/en#/F80 Reference for all ICD-10 disorders mentioned in the table. ^ http://behavenet.com/apa-diagnostic-classification-dsm-iv-tr#301 Reference for all DSM-IV-TR disorders mentioned in the table. ^ https://icd.who.int/browse11/l-m/en#/http%3a%2f%2fid.who.int%2ficd%2fentity%2f334423054 Reference for all ICD-11 disorders mentioned in the table External links [ edit ] Classification D ICD - 10 : F80 , F81 , F82 , F83 ICD - 9-CM : 307 , 315 v t e Dyslexia and related specific developmental disorders Conditions Speech, language , and communication Expressive language disorder Infantile speech Landau–Kleffner syndrome Language disorder Lisp Mixed receptive-expressive language disorder Specific language impairment Speech and language impairment Speech disorder Speech error Speech sound disorder Stuttering Tip of the tongue Learning disability Dyslexia Dyscalculia Dysgraphia Disorder of written expression Motor Developmental coordination disorder Developmental verbal dyspraxia Sensory Auditory processing disorder Sensory processing disorder Related topics Dyslexia research Irlen filters Learning Ally Learning problems in childhood cancer Literacy Management of dyslexia Multisensory integration Neuropsychology Reading acquisition Spelling Writing system Lists Dyslexia in fiction Languages by Writing System People with dyslexia