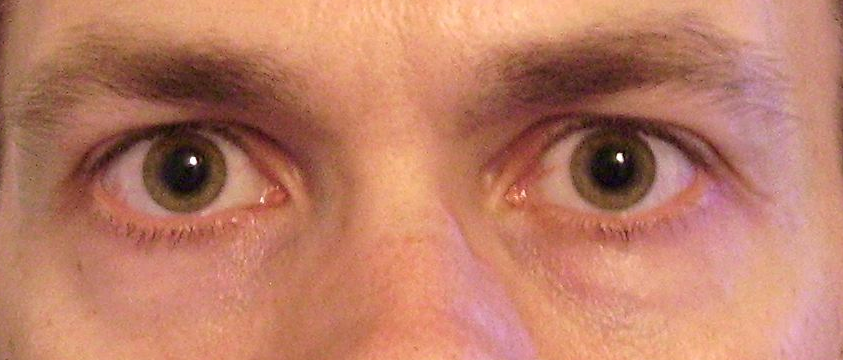
Most women with polycystic ovary syndrome produce excess male sex hormones (androgens), a condition called hyperandrogenism. ... About half of all women with polycystic ovary syndrome are overweight or obese and are at increased risk of a fatty liver. ... Frequency Polycystic ovary syndrome is the most common cause of infertility due to absent ovulation. ... Common variations (polymorphisms) in several genes have been associated with the risk of developing polycystic ovary syndrome. Because they are common, these variations can be present in people with polycystic ovary syndrome and in those without. ... It is estimated that 20 to 40 percent of women with polycystic ovary syndrome have an affected mother or sister.
Overview Polycystic ovary syndrome (PCOS) is a problem with hormones that happens during the reproductive years. ... Complications Complications of PCOS can include: Infertility Gestational diabetes or pregnancy-induced high blood pressure Miscarriage or premature birth Nonalcoholic steatohepatitis — a severe liver inflammation caused by fat buildup in the liver Metabolic syndrome — a cluster of conditions including high blood pressure, high blood sugar, and unhealthy cholesterol or triglyceride levels that significantly increase your risk of heart and blood vessel (cardiovascular) disease Type 2 diabetes or prediabetes Sleep apnea Depression, anxiety and eating disorders Cancer of the uterine lining (endometrial cancer) Obesity commonly occurs with PCOS and can worsen complications of the disorder. Diagnosis There's no single test to specifically diagnose polycystic ovary syndrome (PCOS). Your health care provider is likely to start with a discussion of your symptoms, medications and any other medical conditions.
They examined 115 sisters of 80 probands with PCO syndrome from unrelated families. PCO syndrome was diagnosed by the combination of elevated serum androgen levels and 6 or fewer menses per year with the exclusion of secondary causes. The diagnostic criteria for PCO syndrome were fulfilled in 22% of the sisters. ... The familial aggregation of hyperandrogenemia in PCO syndrome kindreds suggested that it is a genetic trait which can be used to assign affected status in linkage studies designed to identify PCO syndrome genes. Polycystic ovary syndrome is associated with an increased risk of type II diabetes mellitus. ... The pathogenesis of polycystic ovary syndrome was discussed by McKenna (1988) and Givens (1988).
The signs and symptoms of this condition vary widely, even among members of the same family. Branchiootic (BO) syndrome includes many of the same features as BOR syndrome, but affected individuals do not have kidney abnormalities. ... "Oto-" and "-otic" refer to the ear ; most people with BOR/BO syndrome have hearing loss and other ear abnormalities. ... "Renal" refers to the kidneys ; BOR syndrome (but not BO syndrome) causes abnormalities of kidney structure and function. ... Frequency Researchers estimate that BOR/BO syndrome affects about 1 in 40,000 people. ... The resulting genetic changes affect the development of organs and tissues before birth, which leads to the characteristic features of BOR/BO syndrome. Some people with BOR/BO syndrome do not have an identified mutation in any of the genes listed above.
Description Individuals with the BO syndrome are affected by the same branchial and otic anomalies as those seen in individuals with the branchiootorenal syndrome (see BOR1, 113650), but lack renal anomalies (Vincent et al., 1997). Although Melnick et al. (1978) maintained that the BO syndrome is distinct from the BOR syndrome because of the lack of renal anomalies and variable presence of deafness in the former, Cremers and Fikkers-van Noord (1980) suggested that the 2 syndromes represent a single entity. ... Wildervanck (1962) suggested this was a different syndrome from that described by Fourman and Fourman (1955). ... Spruijt et al. (2006) emphasized the phenotypic variability of BO syndrome associated with EYA1 mutations. ... Haan et al. (1989) concluded that the BOS syndrome is caused by mutation at either 8q13.3 or 8q21.13.
A number sign (#) is used with this entry because branchiootic syndrome-3 is caused by mutation in the SIX1 gene (601205). For a phenotypic description and a discussion of genetic heterogeneity of branchiootic syndrome, see BOS1 (602588). Mapping Ruf et al. (2003) mapped a locus for branchiootic syndrome to 14q21.3-q24.3, which they designated BOS3. ... The finding of lacrimal duct stenosis, a common association in both branchiootorenal (BOR; 113650) and branchiootic syndromes, further confirmed the diagnosis of BOS. ... By direct sequencing of exons in these genes, they identified 2 different mutations in the SIX1 gene in 3 kindreds with branchiootic syndrome-3 (601205.0001-601205.0002). In 7 affected individuals from a large 5-generation Danish family with branchiootic syndrome, Sanggaard et al. (2007) identified heterozygosity for a missense mutation in the SIX1 gene (601205.0004).
Hepatopulmonary syndrome Specialty Gastroenterology In medicine , hepatopulmonary syndrome is a syndrome of shortness of breath and hypoxemia (low oxygen levels in the blood of the arteries) caused by vasodilation (broadening of the blood vessels) in the lungs of patients with liver disease . ... Diagnosis [ edit ] The hepatopulmonary syndrome is suspected in any patient with known liver disease who reports dyspnea (particularly platypnea ). ... "Hepatopulmonary Syndrome — A Liver-Induced Lung Vascular Disorder". ... International Liver Transplant Society Practice Guidelines: Diagnosis and Management of Hepatopulmonary Syndrome and Portopulmonary Hypertension. ... "Natural history of hepatopulmonary syndrome: impact of liver transplantation".
Overview Hepatopulmonary (hep-uh-toe-POOL-moe-nar-e) syndrome is an uncommon condition that affects the lungs of people with advanced liver disease. Hepatopulmonary syndrome is caused by blood vessels in the lungs expanding (dilating) and increasing in number, making it hard for red blood cells to properly absorb oxygen. ... A liver transplant is the only cure for hepatopulmonary syndrome. Symptoms Most people with hepatopulmonary syndrome have no symptoms. ... What causes this abnormality remains unclear, and it's unknown why some people with liver disease develop hepatopulmonary syndrome while others do not. Diagnosis These tests can help determine if you have hepatopulmonary syndrome: Pulse oximetry. ... A liver transplant is the only cure for hepatopulmonary syndrome. Clinical trials Explore Mayo Clinic studies testing new treatments, interventions and tests as a means to prevent, detect, treat or manage this condition.
Hepatopulmonary syndrome (HPS) is a lung disease characterized by widening of arteries and veins (dilatation) in the lungs in people who have chronic liver disease .
Short QT syndrome is a condition that can cause a disruption of the heart's normal rhythm (arrhythmia). ... However, some people with short QT syndrome never experience any health problems associated with the condition. Frequency Short QT syndrome appears to be rare. At least 70 cases have been identified worldwide since the condition was discovered in 2000. ... Learn more about the genes associated with Short QT syndrome CACNA1C KCNH2 KCNJ2 KCNQ1 Inheritance Pattern Short QT syndrome appears to have an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Some affected individuals have a family history of short QT syndrome or related heart problems and sudden cardiac death.
A number sign (#) is used with this entry because of evidence that short QT syndrome-3 (SQT3) is caused by heterozygous mutation in the KCNJ2 gene (600681) on chromosome 17q24. Description Short QT syndrome is a cardiac channelopathy associated with a predisposition to atrial fibrillation and sudden cardiac death. ... For a discussion of genetic heterogeneity of short QT syndrome, see SQT1 (609620). Clinical Features Priori et al. (2005) reported an asymptomatic 5-year-old girl who was found to have an abnormal ECG on routine clinical evaluation, with a markedly short repolarization time and conspicuously narrow and peaked T waves (QTc interval, 315 ms). ... Molecular Genetics In a 5-year-old girl and her father who had short QT intervals and asymmetric T waves on ECG and who were negative for mutations in the 2 genes previously associated with short QT syndrome, KCNH2 and KCNQ1, Priori et al. (2005) identified a heterozygous missense mutation in the KCNJ2 gene (600681.0010). ... Schimpf et al. (2005) reviewed the clinical, electrophysiologic, and molecular features of 15 reported cases and 2 unpublished cases of short QT syndrome types 1, 2 (609621), and 3.
A number sign (#) is used with this entry because of evidence that short QT syndrome-1 (SQT1) is caused by heterozygous mutation in the KCNH2 gene (152427) on chromosome 7q36. Description Short QT syndrome is a cardiac channelopathy associated with a predisposition to atrial fibrillation and sudden cardiac death. ... Genetic Heterogeneity of Short QT Syndrome Short QT syndrome-2 (SQT2; 609621) is caused by mutation in the KCNQ1 gene (607542). ... The occurrence of sudden cardiac death in the first 12 months of life in 2 patients suggested the possibility of a link between KCNH2 gain-of-function mutations and sudden infant death syndrome (272120). In a family with short QT syndrome, originally reported by Gussak et al. (2000), Hong et al. (2005) identified an N588K mutation (152427.0017) in the KCNH2 gene. They concluded that codon 588 is a hotspot for this familial form of short QT syndrome. Hong et al. (2005) noted that the disease is clinically heterogeneous, with symptoms varying from atrial to ventricular fibrillation and sudden death in the 3 families with the same mutation.
Epidemiology This extremely rare syndrome affects mainly young adults or infants and has been reported in nearly 80 families. ... Differential diagnosis Differential diagnosis includes other repolarizing disorders such as Brugada syndrome and early repolarization syndrome. Of note, a few rare patients with Brugada syndrome have been reported to have a short QTc.
If affected individuals take this medication, it can cause life-threatening liver failure. Cockayne syndrome is sometimes divided into types I, II, and III based on the severity and age of onset of symptoms. However, the differences between the types are not always clear-cut, and some researchers believe the signs and symptoms reflect a spectrum instead of distinct types. Cockayne syndrome type II is also known as cerebro-oculo-facio-skeletal (COFS) syndrome, and while some researchers consider it to be a separate but similar condition, others classify it as part of the Cockayne syndrome disease spectrum. Frequency Cockayne syndrome is estimated to occur in 2 to 3 per million newborns in the United States and Europe. Causes Cockayne syndrome can result from mutations in either the ERCC6 gene (also known as CSB ) or the ERCC8 gene (also known as CSA ). ... However, in people with Cockayne syndrome, DNA damage is not repaired normally.
Cerebro-oculo-facio-skeletal (COFS) syndrome is a degenerative disorder that primarily involves the brain, eyes, and spinal cord. Affected individuals have mild to severe intellectual disability, severely reduced muscle tone (hypotonia), impaired reflexes, vision impairment, and involuntary eye movements. Children with COFS syndrome have distinctive facial features, including low-set ears, small eyes, small head size (microcephaly), and a small jaw (micrognathia). They may also have abnormalities of the skull, limbs, heart, and kidneys. Individuals with COFS syndrome are often diagnosed at birth. In many cases, the cause of the disorder is unknown. ... When an individual has the features of COFS syndrome and a mutation in the ERCC6 gene, they are said to have Cockayne syndrome type II . COFS syndrome is inherited in an autosomal recessive manner.
Cerebrooculofacioskeletal (COFS) syndrome is a rare genetic disorder, belonging to a family of diseases of DNA repair, characterized by a severe sensorineural involvement. ... To date, fewer than 20 cases have been confirmed, at a cellular or molecular level, as being truly similar to the primary cases described by Lowry, Pena and Shokeir in the indigenous population of Manitoba. Clinical description COFS syndrome constitutes the prenatal extreme form of Cockayne syndrome (see this term). ... Differential diagnosis Differential diagnoses include infectious fetopathies (cytomegalovirus, rubella, toxoplasmosis; see these terms) and MICRO syndrome (see this term) that can present as clinically similar to COFS syndrome, but with normal DNA repair. ... It is confirmed by examination of DNA repair in chorionic villi or amniotic cells and by checking for mutations. Genetic counseling COFS syndrome is transmitted in an autosomal recessive manner. ... Enteric feeding is often necessary. Prognosis COFS syndrome is a severe disease leading to death in the first years of live, particularly by respiratory infections.
Cooks syndrome Other names Anonychia-onychodystrophy with hypoplasia or absence of distal phalanges syndrome Autosomal dominant is the manner of inheritance of this condition Cooks syndrome is a hereditary disorder which is characterized in the hands by bilateral nail hypoplasia on the thumb , index finger , and middle finger , absence of fingernails ( anonychia ) on the ring finger and little finger , lengthening of the thumbs, and bulbousness of the fingers. ... Because the disorder primarily affected the nails and distal phalanges, the research group concluded that brachydactyly type B and Cooks syndrome are the same disorder. [3] However, in 2007, a 2-year-old girl was found with symptoms consistent with both brachydactyly type B and Cooks syndrome. ... "Anonychia and absence/hypoplasia of distal phalanges (Cooks syndrome): report of a second family" . ... (January 1985). "A new nail dysplasia syndrome with onychonychia and absence and/or hypoplasia of distal phalanges". ... "Brachydactyly type B with its distinct facies and 'Cooks syndrome' are the same entity". Clinical Dysmorphology . 8 (1): 41–5. doi : 10.1097/00019605-199901000-00008 .
Generally the nails of the first to third digits are progressively deformed with total anonychia in the last 2 digits and in all toes (summary by Genzer-Nir et al., 2010). A syndrome has been described in which affected females display juvenile hypertrophy of the breast (JHB; 113670) in association with ODP, whereas males have only ODP (mammary-digital-nail syndrome; 613689). ... De Ravel et al. (1999) reported a sib pair with a distinct facies, absence/hypoplasia of the distal phalanges of the hands, feet, and nails, and bulbous digit tips, and suggested that these patients had features of both classic type B brachydactyly (BDB; 113000) and Cooks syndrome. Castori et al. (2007) reported a 2-year-old Caucasian girl with Cooks syndrome. ... Castori et al. (2007) concluded that Cooks syndrome is distinct from classic brachydactyly type B because the former affects dorsoventral patterning and the latter affects early differentiation of skeletal precursor structures. The authors suggested that the large family reported by Kumar and Levick (1986) (106990) may have had Cooks syndrome. Kurth et al. (2009) reported 4 families with symmetric brachydactyly of the hands and feet, along with hyponychia or anonychia. The phenotype was consistent with Cooks syndrome. Radiographs showed missing middle phalanges and elongated terminal and proximal phalanges.
Cooks syndrome is a malformation syndrome affecting the apical structures of digits and presenting with hypo/aplasia of nails and distal phalanges. ... Epidemiology To date, less than 20 individuals have been described in the world literature. Clinical description Cooks syndrome is congenital and presents with hypo/anonychia, small or absent distal phalanges and digitalization of the thumbs. ... To date, no facial dysmorphism has been associated with Cooks syndrome. Cooks syndrome is considered a clinical form of brachydactyly type B (see this term), distinct from the typical variant with sparing or duplication of the thumbs and caused by mutations in the ROR2 gene (9q22). Etiology The exact etiology of Cooks syndrome is still unknown but microduplications on chromosome 17q24.3 and involvement of a non-coding element of the SOX9 gene (17q24.3) have recently been associated in some patients with Cooks syndrome.
Trichohepatoenteric syndrome is a condition that affects the hair (tricho-), liver (hepato-), and intestines (enteric), as well as other tissues and organs in the body. This condition is also known as syndromic diarrhea because chronic, difficult-to-treat diarrhea is one of its major features. ... Most children with trichohepatoenteric syndrome are small at birth, and they remain shorter than their peers throughout life. ... Less commonly, trichohepatoenteric syndrome is associated with heart (cardiac) abnormalities. ... Frequency Trichohepatoenteric syndrome is a rare condition with an estimated prevalence of about 1 in 1 million people.
Arts syndrome is a disorder that causes serious neurological problems in males. ... Frequency Arts syndrome appears to be extremely rare. Only a few families with this disorder have been described in the medical literature. ... The PRPS1 gene mutations that cause Arts syndrome replace single protein building blocks (amino acids ) in the PRPP synthetase 1 enzyme. ... Impairment of these processes may have a particularly severe effect on tissues that require a large amount of energy, such as the nervous system, resulting in the neurological problems characteristic of Arts syndrome. The reason for the increased risk of respiratory infections in Arts syndrome is unclear. Learn more about the gene associated with Arts syndrome PRPS1 Inheritance Pattern This condition is inherited in an X-linked pattern.
Twelve of 15 boys from the two Dutch families reported with Arts syndrome died before age six years of complications of infection. ... Surveillance: Regular neuropsychological, audiologic, and ophthalmologic examinations. Genetic counseling. Arts syndrome is inherited in an X-linked manner. ... Diagnosis Formal diagnostic criteria for Arts syndrome have not been established. Suggestive Findings Arts syndrome, part of the spectrum of PRPS1 -related disorders, should be suspected in a male proband with the following clinical and laboratory features. ... Prevalence Four kindreds with Arts syndrome have been identified worldwide [de Brouwer et al 2007, Synofzik et al 2014, Maruyama et al 2016]. ... Mildly affected carrier females from families with Arts syndrome may also benefit from SAM supplementation in their diet, although this remains to be tested.
Arts syndrome Other names ataxia-deafness-optic atrophy, lethal; ataxia - fatal x-linked with deafness and loss of vision This condition is inherited in an X-linked recessive manner Arts syndrome is a rare metabolic disorder that causes serious neurological problems in males due to a malfunction of the PRPP synthetase 1 enzyme. ... Ile275Thr and p.Gly306Glu [8] Genetics [ edit ] Arts syndrome follows an X-linked inheritance. ... Only three families with CMTX5 and two families Arts syndrome, respectively, have been reported worldwide so far. ... Sequence analysis of PRPS1, the only gene associated with Arts syndrome, has detected mutations in both kindreds reported to date. ... "X-linked Charcot-Marie-Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: evidence from a family with a novel PRPS1 mutation" .
Lethal ataxia with deafness and optic atrophy (also known as Arts syndrome) is characterized by intellectual deficit, early-onset hypotonia, ataxia, delayed motor development, hearing impairment and loss of vision due to optic atrophy. Epidemiology It was initially described in 12 male members from five generations of a Dutch family. Arts syndrome has also been described in one Australian family. ... Etiology It is caused by missense mutations in the phosphoribosyl pyrophosphate synthetase 1 gene ( PRPS1 ) localized to Xq22.1-q24, leading to impaired purine biosynthesis. Genetic counseling Arts syndrome is transmitted as an X-linked recessive trait.
A number sign (#) is used with this entry because of evidence that Arts syndrome (ARTS) is caused by loss-of-function mutation in the PRPS1 gene (311850) on chromosome Xq22. ... There is considerable phenotypic overlap between Arts syndrome, CMTX5, and DFNX1, as well as intrafamilial variability depending on gender, X-inactivation ratio, residual enzyme activity, and additional factors. ... In addition, he had recurrent infections and early death at age 27 months from infection, consistent with Arts syndrome. A maternal uncle with similar symptoms had died of pneumonia at age 2. ... Mapping Using linkage analysis, Kremer et al. (1996) localized the gene (or genes) responsible for the Arts syndrome phenotype to Xq21.33-q24 between DXS1231 and DXS1001 with a maximum lod score of 6.97. ... De Brouwer et al. (2010) reported preliminary results of the 2 Australian brothers with Arts syndrome, which revealed some improvement of their condition.
Arts syndrome is characterized by sensorineural hearing loss and serious neurological and immune system problems in males. Females can also be affected by this condition, but they typically have much milder symptoms. Arts syndrome is caused by mutations in the PRPS1 gene which is located on the X chromosome.
Oculofaciocardiodental (OFCD) syndrome is a condition that affects the development of the eyes (oculo-), facial features (facio-), heart (cardio-) and teeth (dental). ... Frequency OFCD syndrome is very rare; the incidence is estimated to be less than 1 in 1 million people. Causes Mutations in the BCOR gene cause OFCD syndrome. The BCOR gene provides instructions for making a protein called the BCL6 corepressor. ... Learn more about the gene associated with Oculofaciocardiodental syndrome BCOR Inheritance Pattern This condition is inherited in an X-linked dominant pattern . ... A lack of this protein appears to be lethal very early in development, so no males are born with OFCD syndrome.
Oculo-facio-cardio-dental syndrome (OFCD) is a very rare multiple congenital anomaly syndrome characterized by dental radiculomegaly, congenital cataract, facial dismorphism and congenital heart disease. ... Diagnostic methods Diagnosis of OFCD is hard for medical specialists and the syndrome is often unrecognized. The specific dental findings (visible on a panoramic radiograph of the jaws) can be easily diagnosed by an orthodontist or a dentist. Genetic counseling OFCD is an X-linked dominant syndrome that is lethal in males. Management and treatment Management requires appropriate cardiac, ophthalmic and dental care.
Oculofaciocardiodental syndrome is a genetic syndrome that affects the eyes, heart, face, and teeth. ... Eye symptoms may involve one or both eyes.Oculofaciocardiodental syndrome is caused by mutations in the BCOR gene and is inherited in an X-linked dominant fashion .
Oculofaciocardiodental syndrome This condition is inherited in an X-linked dominant manner. Specialty Medical genetics Oculofaciocardiodental syndrome is a rare X-linked dominant genetic disorder . [1] Contents 1 Presentation 2 Genetics 3 Diagnosis 4 Treatment 5 History 6 References Presentation [ edit ] The incidence of this condition is less than 1 per million. ... A genetically related disorder is Lenz microphthalmia syndrome . [2] Diagnosis [ edit ] This section is empty. ... "Oculofaciocardiodental syndrome: novel BCOR mutations and expression in dental cells" . ... "A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome" . J. Med. Genet . 51 (3): 185–96. doi : 10.1136/jmedgenet-2013-101660 .
Hyperparathyroidism-jaw tumor syndrome is a condition characterized by overactivity of the parathyroid glands (hyperparathyroidism). ... In people with hyperthyroidism-jaw tumor syndrome, hyperparathyroidism is caused by tumors that form in the parathyroid glands. ... Approximately 15 percent of people with hyperparathyroidism-jaw tumor syndrome develop a cancerous tumor called parathyroid carcinoma. People with hyperparathyroidism-jaw tumor syndrome may also have a type of benign tumor called a fibroma in the jaw. ... Some people with hyperparathyroidism-jaw tumor syndrome do not have identified mutations in the CDC73 gene.
A number sign (#) is used with this entry because hyperparathyroidism-2 with jaw tumors, also known as hyperparathyroidism-jaw tumor syndrome, is caused by heterozygous mutation in the CDC73 gene (607393) on chromosome 1q32. Description Hyperparathyroidism-jaw tumor syndrome is a rare autosomal dominant disorder characterized by synchronous or metachronous occurrence of primary hyperparathyroidism, ossifying fibroma of the maxilla and/or mandible, renal tumor, and uterine tumors. ... The occurrence of Wilms tumor in 2 female members of unrelated families in their study raised the possibility that Wilms tumor may be a component of the HPT-JT syndrome. Further evidence that parathyroid carcinoma and Wilms tumor are part of the HPT-JT syndrome came from a report by Kakinuma et al. (1994) in which one sib had parathyroid carcinoma, a second had parathyroid adenoma plus Wilms tumor, and a third had parathyroid adenoma plus jaw tumor. ... Teh et al. (1996) reported 2 families with HPT-JT syndrome in which adult renal hamartomas or cystic kidney disease were prominent associated features, possibly representing a new phenotypic variant of the HPT-JT syndrome. ... This and other experiences established parathyroid cancer as part of the hyperparathyroidism-jaw tumor syndrome occurring in at least 1 patient in 5 (42%) of the 12 known families.
A rare genetic disease characterized by synchronous or metachronous occurrence of primary hyperparathyroidism and ossifying fibroma of the maxilla and/or mandible, associated with an increased risk of parathyroid carcinoma. Occurrence of renal cysts or tumors, multiple uterine polyps, and thyroid tumors has also been reported.
Hyperparathyroidism-jaw tumor syndrome (HPT-JT) is an inherited condition that causes overactivity of the parathyroid glands ( hyperparathyroidism ). These glands regulate the body's use of calcium, so overactivity can lead to high calcium levels in the blood ( hypercalcemia ). The syndrome typically begins in late adolescence or early adulthood.
Description Asperger syndrome is considered to be a form of childhood autism (see, e.g., 209850). The DSM-IV (American Psychiatric Association, 1994) specifies several diagnostic criteria for Asperger syndrome, which has many of the same features as autism. ... Gillberg et al. (2001) described the development of the Asperger syndrome (and high-functioning autism) Diagnostic Interview (ASDI), which they claimed has a strong validity in the diagnosis of the disorder. For a discussion of genetic heterogeneity of Asperger syndrome, see ASPG1 (608638). Mapping Ylisaukko-oja et al. (2004) performed a genomewide scan on 17 Finnish families ascertained for Asperger syndrome with a strictly defined phenotype. ... Rehnstrom et al. (2006) enlarged the study of Asperger syndrome in Finnish families by analyzing an independent set of 12 novel extended Asperger families with 54 individuals having Asperger syndrome or Asperger-like symptoms.
Adie's syndrome Other names Holmes–Adie syndrome, Adie's tonic pupil, Holmes–Adie pupil Bilateral mydriasis given the observational diagnosis Adie's pupils by an ophthalmologist Pronunciation / ˈ eɪ d i / Specialty Ophthalmology Adie syndrome , also known as Holmes-Adie syndrome , is a neurological disorder characterized by a tonically dilated pupil that reacts slowly to light but shows a more definite response to accommodation (i.e., light-near dissociation). [1] It is frequently seen in females with absent knee or ankle jerks and impaired sweating. ... Additionally, patients with Holmes-Adie Syndrome can also experience problems with autonomic control of the body. ... See also [ edit ] Ciliary ganglion Ross' syndrome References [ edit ] ^ a b c d e f g h i j "Holmes-Adie syndrome Information Page" . ... "William John Adie: the man behind the syndrome" (PDF) . Clinical & Experimental Ophthalmology . 42 (8): 778–84. doi : 10.1111/ceo.12301 . ... ISBN 978-0-443-06603-0 . ^ "Diagnosis of Adie syndrome WrongDiagnosis.com" . Retrieved 2008-01-21 .
In some patients, patchy hypo- or anhidrosis may also be present (a variant known as Ross syndrome). The condition typically occurs in young adults, with a female preponderance.
From electrophysiologic studies carried out in 11 patients with Adie syndrome, Miyasaki et al. (1988) concluded that the hyporeflexia in this condition is due to the loss of large spindle afferents or the reduced effectiveness of their monosynaptic connections to motoneurons. ... They noted that when diminished deep tendon reflexes are associated with a tonic pupil, the condition is designated Holmes-Adie syndrome. Misc - Stationary, harmless disorder Eyes - Sluggish pupillary response - Mecholyl-sensitive pupil Neuro - Hyporeflexia Inheritance - Autosomal dominant ▲ Close
Adie syndrome is is a neurological disorder affecting the pupil of the eye and the autonomic nervous system . ... In most cases, the cause of Adie syndrome is unknown. Some cases may result from trauma, surgery, lack of blood flow, or infection. ... Glasses and eye drops may help when treatmend is needed. The term Adie syndrome is used when both the pupil and deep tendon reflexes are affected.
Mosaic trisomy 16 is a rare chromosomal anomaly syndrome with a highly variable phenotype ranging from minor anomalies with normal development to intrauterine growth retardation, abnormal skin pigmentation, craniofacial and body asymmetry, cardiac (e.g. ventricular septal defect) and genital (e.g. hypospadias, cryptorchidism) anomalies, scoliosis and hearing loss to neonatal death.
A phenotypic variant of Bartter syndrome presenting antenatally with maternal polyhydramnios, pre-term delivery and postnatally with polyuria, and nephrocalcinosis. ... Genotypically they comprise Type 1 and Type 2 Bartter syndrome Epidemiology Prevalence of antenatal Bartter syndrome is not exactly known but it comprises about half the cases of Bartter syndrome. Clinical description Typically, antenatal Bartter syndrome manifests prenatally with maternal polyhydramnios (due to fetal polyuria) usually evident by the end of second trimester, often leading to preterm labour and prematurity. ... Patients with type 2 genotype present with a transient hyperkalemic acidosis in the neonatal period; they later manifest with a less severe hypokalemic alkalosis. Etiology Type 1 Bartter syndrome is caused by mutations in the SLC12A1 (15q15-q21) encoding sodium-potassium-chloride co-transporter protein, NKCC2 whereas Type 2 Bartter syndrome is caused by mutations in KCNJ1 (11q24) encoding renal outer medullary potassium channel, ROMK.
Wernicke-Korsakoff syndrome is a brain disorder, due to thiamine deficiency that has been associated with both Wernicke's encephalopathy and Korsakoff syndrome . The term refers to two different syndromes, each representing a different stage of the disease. Wernicke's encephalopathy represents the "acute" phase and Korsakoff's syndrome represents the "chronic" phase. ... Wernicke's encephalopathy is characterized by confusion, abnormal stance and gait (ataxia), and abnormal eye movements (nystagmus). Korsakoff's syndrome is observed in a small number of patients. ... Wernicke encephalopathy is an acute syndrome and requires emergency treatment to prevent death and neurologic complications.
Wernicke-Korsakoff syndrome is characterized by acute encephalopathy followed by chronic impairment of short-term memory. ... The isozyme pattern found in 39 of 42 patients with Wernicke-Korsakoff syndrome was present in only 8 of 36 controls. ... Probably this means that the Wernicke-Korsakoff syndrome is a recessive disorder, presumably autosomal recessive. Two of the patients with the syndrome whose cells were studied by Blass and Gibson (1977) were female. ... The syndrome is said to be rare in American blacks.
Mental illness caused by a lack of thiamine in the brain See also: Wernicke–Korsakoff syndrome Korsakoff syndrome Other names Alcoholic Korsakoff syndrome (AKS), Korsakov syndrome, Alcohol amnestic disorder Thiamine Specialty Psychiatry Korsakoff syndrome ( KS ) [1] is an amnestic disorder caused by thiamine (vitamin B 1 ) deficiency typically associated with prolonged use of alcohol . [2] The syndrome and psychosis are named after Sergei Korsakoff , the Russian neuropsychiatrist who discovered it during the late 19th century. ... When Wernicke encephalopathy accompanies Korsakoff syndrome the combination is called Wernicke–Korsakoff syndrome ; however, a recognized episode of Wernicke encephalopathy is not always obvious. ... Empirical research has suggested that good health practices have beneficial effects in Korsakoff syndrome. [26] Epidemiology [ edit ] Rates varies between country, but it is estimated to affect around 12.5% of heavy drinkers. [28] References [ edit ] ^ "Korsakoff Syndrome - MeSH - NCBI" . www.ncbi.nlm.nih.gov . ^ "Korsakoff syndrome" . ... "Wernicke-Korsakoff syndrome" . MedlinePlus Medical Encyclopedia . ... "Prevalence of Wernicke-Korsakoff syndrome in Australia: has thiamine fortification made a difference?"
Ataxia-pancytopenia syndrome Specialty Neurology Ataxia-pancytopenia syndrome is a rare autosomal dominant disorder characterized by cerebellar ataxia , peripheral neuropathies , pancytopenia and a predilection to myelodysplastic syndrome and acute myeloid leukemia . Contents 1 Genetics 2 Diagnosis 3 History 4 References Genetics [ edit ] This syndrome is caused by mutations in the sterile alpha motif domain containing 9-like ( SAMD9L ) gene. [1] This gene is located on the long arm of chromosome 7 . ... You can help by adding to it . ( July 2018 ) History [ edit ] This syndrome was first described in 1981. [2] References [ edit ] ^ Chen DH, Below JE, Shimamura A, Keel SB, Matsushita M, Wolff J, Sul Y, Bonkowski E, Castella M, Taniguchi T, Nickerson D, Papayannopoulou T, Bird TD, Raskind WH (2016) Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet 98(6):1146-1158. doi: 10.1016/j.ajhg.2016.04.009 ^ Li FP, Hecht F, Kaiser-McCaw B, Baranko PV, Potter NU (1981) Ataxia-pancytopenia: syndrome of cerebellar ataxia, hypoplastic anemia, monosomy 7, and acute myelogenous leukemia.
A number sign (#) is used with this entry because of evidence that ataxia-pancytopenia syndrome (ATXPC) is caused by heterozygous mutation in the SAMD9L gene (611170) on chromosome 7q21. Description Ataxia-pancytopenia syndrome is an autosomal dominant disorder characterized by cerebellar ataxia, variable hematologic cytopenias, and presdisposition to bone marrow failure and myeloid leukemia (summary by Chen et al., 2016) Clinical Features Li et al. (1978, 1981) described a family with ataxia and pancytopenia. ... Chen et al. (2016) reported a large multigenerational family (UW-AP) of German, Irish, and Native American descent in which at least 9 individuals had ataxia-pancytopenia syndrome. The phenotype was highly variable, with onset of symptoms ranging from 6 to 62 years of age, although manifestations could be detected earlier by examination. ... The phenotype was considered consistent with the ataxia-pancytopenia syndrome, although monosomy of chromosome 7 was not found in bone marrow.
Ataxia-pancytopenia syndrome is a rare condition that affects the part of the brain that coordinates movement (the cerebellum) and blood-forming cells in the bone marrow . ... People with ataxia-pancytopenia syndrome have neurological problems associated with a loss of tissue (atrophy) and other changes in the cerebellum. ... Frequency Ataxia-pancytopenia syndrome appears to be very rare. At least 25 affected individuals from four families have been described in the medical literature. Causes Ataxia-pancytopenia syndrome is caused by inherited mutations in the SAMD9L gene. ... The mutations that cause ataxia-pancytopenia syndrome are described as "gain-of-function."
A rare genetic disease characterized by cerebellar ataxia, cytopenias and predisposition to bone marrow failure and myeloid leukaemia. Neurologic features variably include slowly progressive cerebellar ataxia or balance impairment with cerebellar atrophy and periventricular white matter T2 hyperintensities in brain MRI, horizontal and vertical nystagmus, dysmetria, dysarthria, pyramidal tract signs and reduced nerve conduction velocity. Hematological abnormalities are variable and may be intermittent and include cytopenias of all cell lineages, immunodeficiency, myelodysplasia and acute myeloid leukemia.
A possible relationship to Chediak-Higashi syndrome (214500) was unclear. The phenotype is also reminiscent of the Cross oculocerebral syndrome (257800). ... Duran-McKinster et al. (1999) recommended that the differential diagnosis include Chediak-Higashi syndrome and Griscelli syndrome (214450). ... Molecular Genetics Sanal et al. (2000) referred to neuroectodermal melanolysosomal disease as an allelic variant of Griscelli syndrome. Anikster et al. (2002) suggested that families previously thought to have Griscelli syndrome due to mutations in the MYO5A gene (160777) may in fact have suffered from Elejalde syndrome, the lack of immunologic defects being an important distinction from Griscelli syndrome (see Griscelli syndrome type 2, 607624). Menasche et al. (2002), Huizing et al. (2002), and Bahadoran et al. (2003, 2003) also suggested the identity of Elejalde syndrome, at least in some patients, and Griscelli syndrome type 1. ... By contrast, the second series of Elejalde syndrome patients (Duran-McKinster et al., 1999; Ivanovich et al., 2001) had clinical and histologic features suggestive of Griscelli syndrome type 1.
Griscelli syndrome is an inherited condition characterized by unusually light (hypopigmented) skin and light silvery-gray hair starting in infancy. ... Another condition called Elejalde disease has many of the same signs and symptoms, and some researchers have proposed that Griscelli syndrome type 1 and Elejalde disease are actually the same disorder. People with Griscelli syndrome type 2 have immune system abnormalities in addition to having hypopigmented skin and hair. ... Unusually light skin and hair coloring are the only features of Griscelli syndrome type 3. People with this form of the disorder do not have neurological abnormalities or immune system problems. Frequency Griscelli syndrome is a rare condition; its prevalence is unknown.
An extremely rare lethal autosomal recessive disorder characterized by massive birth weight, swollen globular body, generalized edema, short limbs, postaxial polydactyly, thick skin, facial dysmorphism (slanted palpebral fissures, hypertelorism, epicanthic folds, dysplastic ears), excessive connective tissue, renal dysplasia, and in some patients, organomegaly, craniosynostosis with acrocephaly, omphalocele, cleft palate, and cryptorchidism. Fewer than 10 cases have been reported to date.
Elejalde syndrome (ES) is characterized by silvery to leaden hair, bronze skin colour in sun-exposed areas and severe neurological impairment. Epidemiology The syndrome was first described in 1979 in three consanguineous families. ... Etiology The etiology of ES is still unknown, but recent molecular data have shed light on the complex relationship that exists between ES and the Griscelli syndrome (see this term). Mutations in the myosin Va gene ( MYOVA ) result in the so-called Griscelli syndrome type 1, characterized by cutaneous and neurologic manifestations. ... It is very likely that Griscelli syndrome type 1 corresponds to ES. Differential diagnosis The main differential diagnoses are Griscelli syndrome type 2 (caused by mutations in RAB27A ), which is characterized by cutaneous and immunological manifestations, without primary neurological signs; Griscelli syndrome type 3, which has only cutaneous hypopigmentation as a feature and is caused by mutations in MLPH ; and Chediak-Higashi syndrome (see this term), also characterized by silvery hair, and by oculocutaneous hypopigmentation.
Description Acrocephalopolydactylous dysplasia, or Elejalde syndrome, is a lethal multiple congenital disorder characterized by increased birth weight, globular body with thick skin, organomegaly, and fibrosis in multiple tissues (summary by Phadke et al., 2011). ... Lurie et al. (1991) reviewed cerebrorenodigital syndromes, of which 19 were considered to be autosomal recessive. Nevin et al. (1994) reported an 18-week-old fetus with craniosynostosis, gross edema, short limbs, postaxial polydactyly, redundant connective tissue, and cystic renal dysplasia consistent with a diagnosis of Elejalde syndrome. Thornton and Stewart (1997) described another example of this disorder in a child born of nonconsanguineous parents. ... Phadke et al. (2011) reported a 33-week-old fetus, born of unrelated Indian parents, with features consistent with Elejalde syndrome. The fetus had a globular external appearance with distended, thick skin, bloated face, and ascites.
Atypical hemolytic-uremic syndrome is a disease that primarily affects kidney function. ... These life-threatening complications prevent the kidneys from filtering fluids and waste products from the body effectively. Atypical hemolytic-uremic syndrome should be distinguished from a more common condition called typical hemolytic-uremic syndrome. ... Frequency The incidence of atypical hemolytic-uremic syndrome is estimated to be 1 in 500,000 people per year in the United States. ... Causes Atypical hemolytic-uremic syndrome often results from a combination of environmental and genetic factors. ... Mutations in the genes associated with atypical hemolytic-uremic syndrome lead to uncontrolled activation of the complement system.
Typical hemolytic-uremic syndrome (typical HUS) is a thrombotic microangiopathy characterized by mechanical hemolytic anemia, thrombocytopenia, and renal dysfunction that is usually associated with prodromal enteritis caused by Shigella dysentriae type 1 or E.
Hamman's syndrome A CT scan showing air in the mediastinum with subcutaneous emphysema, which can result in Hamman's syndrome Specialty Pulmonology Hamman's syndrome , also known as Macklin's syndrome , is a syndrome of spontaneous subcutaneous emphysema [1] (air in the subcutaneous tissues of the skin) and pneumomediastinum (air in the mediastinum , the center of the chest cavity ), sometimes associated with pain and, less commonly, dyspnea (difficulty breathing), dysphonia , and a low-grade fever . Hamman's syndrome can cause Hamman's sign , an unusual combination of sounds that can be heard with a stethoscope . ... "Spontaneous mediastinal emphysema: hamman's syndrome". Ann Chir Gynaecol Fenn . 64 (1): 44–6. ... "Postpartum pneumomediastinum (Hamman's syndrome)" . CMAJ . 177 (1): 32. doi : 10.1503/cmaj.061581 . ... "Spontaneous pneumomediastinum (Hamman's syndrome)". The Surgeon . 8 (2): 63–66. doi : 10.1016/j.surge.2009.10.007 . ^ synd/3004 at Who Named It?
Genitopatellar syndrome Other names Absent patellae-scrotal hypoplasia-renal anomalies-facial dysmorphism-intellectual disability syndrome [1] Genitopatellar syndrome is a rare disorder with characteristic craniofacial features, congenital flexion contractures of the lower limbs, absent or abnormal patellae, urogenital anomalies, and severe psychomotor retardation. [2] In 2012, it was shown that mutations in the gene KAT6B cause the syndrome. [3] Contents 1 Signs and symptoms 2 Cause 3 Diagnosis 4 Treatment 5 History 6 See also 7 References 8 External links Signs and symptoms [ edit ] Genitopatellar syndrome is characterized by genital abnormalities, missing or underdeveloped kneecaps (patellae), intellectual disability and abnormalities affecting other parts of the body. ... On the other hand, the mutation of KAT6B leading to the specific features of genitopatellar syndrome is still not surely proven. [5] Diagnosis [ edit ] Even though clinical diagnostic criteria have not been 100 percent defined for genitopatellar syndrome, the researchers stated that the certain physical features could relate to KAT6B mutation and result in the molecular genetic testing. ... The condition is now known as genitopatellar syndrome. [6] See also [ edit ] Say-Barber-Biesecker-Young-Simpson syndrome References [ edit ] ^ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Genitopatellar syndrome" . www.orpha.net . Retrieved 15 March 2019 . ^ Penttinen, Maila; Koillinen Hannele; Niinikoski Harri; Mäkitie Outi; Hietala Marja (Mar 2009). " Genitopatellar syndrome in an adolescent female with severe osteoporosis and endocrine abnormalities". ... University of Washington, Seattle. 1993. ^ "Genitopatellar syndrome" . ^ "SpringerReference - Meteor" .
The SBBYS variant of Ohdo syndrome (603736) is an allelic disorder with overlapping features. ... Cormier-Daire et al. (2000) reported the first female patient with genitopatellar syndrome, who had clitoral hypertrophy and prominent labia minora. ... Lammer and Abrams (2002) commented that positioning of the legs with flexion contractures at the hips and knees, with the feet hyperflexed and varus, seemed to be characteristic for the syndrome. Armstrong and Clarke (2002) described a 3-month-old male patient with genitopatellar syndrome, born to nonconsanguineous parents. ... The authors stated that this was the oldest reported patient with genitopatellar syndrome, and suggested that her severe osteoporosis and endocrine abnormalities were manifestations of the syndrome. ... Campeau et al. (2012) noted that mutations in the KAT6B gene can also cause the Say-Barber-Biesecker variant of Ohdo syndrome (SBBYSS; 603736), a disorder with features overlapping those of genitopatellar syndrome, but with clinical differences: structural brain defects, skeletal defects, anogenital anomalies, and renal defects are more severe or frequent in genitopatellar syndrome, whereas ocular, dental, palatal, and thyroid defects are more severe or frequent in SBBYSS.
Genitopatellar syndrome is a rare congenital patellar anomaly syndrome characterized by patellar aplasia or hypoplasia associated with microcephaly, characteristic coarse facial features (microcephaly, bitemporal narrowing, large, broad nose with high nasal bridge, prominent cheeks and micro/retrognathia or prognathism), arthrogryposis of the hips and knees, urogenital abnormalities and intellectual deficiency.
Thyroid problems have been reported in some cases. Genitopatellar syndrome (GPS) is caused by changes or mutations in the KAT6B gene.. ... As of 2016, medical researchers are trying to decide if genitopatellar syndrome (GPS) is a separate syndrome from Say–Barber–Biesecker–Young–Simpson syndrome (SBBYSS). Both syndromes are caused by changes (mutations) in the same gene ( KAT6B ). The two syndromes also share a lot of the same features, and some children with mutations in KAT6B gene have a combination of features that fall in between the GPS and SBBYSS. This suggests that GPS and SBBYSS may be two ends of a spectrum of the same syndrome..
Genitopatellar syndrome is a rare condition characterized by genital abnormalities, missing or underdeveloped kneecaps (patellae), intellectual disability, and abnormalities affecting other parts of the body. ... The condition can also be associated with abnormalities of the heart, kidneys, and teeth. Frequency Genitopatellar syndrome is estimated to occur in fewer than 1 per million people. At least 18 cases have been reported in the medical literature. Causes Genitopatellar syndrome is caused by mutations in the KAT6B gene. ... The mutations that cause genitopatellar syndrome occur near the end of the KAT6B gene and lead to the production of a shortened histone acetyltransferase enzyme. ... However, it is unclear how these changes lead to the specific features of genitopatellar syndrome. Learn more about the gene associated with Genitopatellar syndrome KAT6B Inheritance Pattern This condition has an autosomal dominant inheritance pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder.