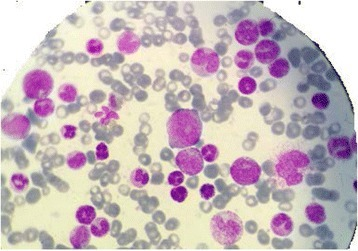
A group of rare immunodeficiency-associated lymphoproliferative disorders characterized by lymphoid or plasmacytic proliferations developing in the context of immunosuppression in a recipient of a solid organ or stem cell allograft. The group includes non-destructive post-transplant lymphoproliferative disorders (PTLDs), polymorphic PTLD, monomorphic PTLDs, and classic Hodgkin lymphoma PTLD. Patients may have more than one type of PTLD in a single or in different locations. The most commonly involved sites are lymph nodes, gastrointestinal tract, lungs, and liver, although the disease may occur almost anywhere in the body. In solid organ transplant recipients, PTLD may also involve the allograft.
Molecular Genetics In a cohort of 38 probands diagnosed with DA2B (Sheldon-Hall syndrome; SHS) who were negative for mutation in the TNNI2 (191043) or TNNT3 (191044) genes, Toydemir et al. (2006) screened the MYH3 gene and identified heterozygous mutations (see, e.g., 160720.0003, 160720.0005, and 160720.0006) in 5 (42%) of 12 familial and 7 (27%) of 26 sporadic cases. Three patients with Freeman-Sheldon syndrome (DA2A; 193700) and 2 patients diagnosed with DA2B shared the T178I mutation in the MYH3 gene (160720.0003).
At age 20 years, she developed nephrotic syndrome after a throat infection. Kidney biopsy showed membranoproliferative glomerulonephritis, and laboratory values showed decreased serum C3 and C4, as well as proteinuria, hematuria, and leukocyturia. ... INHERITANCE - Isolated cases HEAD & NECK Face - Loss of subcutaneous adipose tissue from face - Sunken face - 'Progeroid' expression GENITOURINARY Kidneys - Membranoproliferative glomerulonephritis (26%) - Nephrotic syndrome MUSCLE, SOFT TISSUES - Loss of subcutaneous adipose tissue from face, progressive - Loss of subcutaneous adipose tissue from upper limbs and trunk IMMUNOLOGY - Frequent infections - Decreased serum C3 - Presence of C3 nephritic factor autoantibody LABORATORY ABNORMALITIES - Hematuria - Proteinuria MISCELLANEOUS - Onset in first or second decade - More common in females (male:female ratio 4:1) - Variable phenotype - No family history - Association with autoimmune diseases ▲ Close
Mulibrey nanism Other names Perheentupa syndrome and Pericardial constriction with growth failure [1] Mulibrey nanism has an autosomal recessive pattern of inheritance Specialty Rheumatology , medical genetics Symptoms Infertility [1] Causes Mutation of the TRIM37 gene [2] Diagnostic method Genetic testing [3] Treatment Growth hormone treatment, Regular pelvic exams [4] Mulibrey nanism (" MU scle- LI ver- BR ain- EY e nanism"), is a rare autosomal recessive congenital disorder . ... External links [ edit ] Classification D ICD - 10 : Q87.1 OMIM : 253250 MeSH : D050336 C538604 D050336 DiseasesDB : 32892 External resources Orphanet : 2576 Scholia has a topic profile for Mulibrey nanism . v t e Genetic disorder , organelle: Peroxisomal disorders and lysosomal structural disorders Peroxisome biogenesis disorder Zellweger syndrome Neonatal adrenoleukodystrophy Infantile Refsum disease Adult Refsum disease-2 RCP 1 Enzyme-related Acatalasia RCP 2&3 Mevalonate kinase deficiency D-bifunctional protein deficiency Adult Refsum disease-1 Transporter-related X-linked adrenoleukodystrophy Lysosomal Danon disease See also: proteins , intermediates v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
Hall (1965) described 5 families in which 14 cases of myxedema occurred in addition to the 5 probands. In 1 of these families, a case of thyrotoxicosis was also observed, and in each of 2 families a relative had nontoxic goiter. A sixth proband had a daughter with thyrotoxicosis. In the families of 32 other patients with myxedema, no thyroid dysfunction was detected. Environmental factors, such as viral infection, cannot be excluded in the causation of such familial aggregation. However, the findings were considered compatible with sex-influenced recessive inheritance and also with the previous suggestion of a genetic relationship of myxedema to hyperthyroidism and to nontoxic goiter.
Some have suggested the disease may be a sporadic form of Gerstmann–Sträussler–Scheinker syndrome (GSS). [7] In 2013, Zou W.Q. and coworkers revealed that the peculiar protease-resistant PrP (PrPres) originally found in VPSPr is also detectable in the brain of patients with a genetic CJD linked to PrP Valine (V) to isoleucine (I) mutation at residue 180 (PrPV180I); moreover, they found that the pathological PrP from both VPSPr and gCJDPrPV180I shares a similar glycoform-selective prion formation mechanism.[8,9] Interestingly, the authors further demonstrated that the protease-resistant PrPres from both VPSPr and gCJDV180I lacks the PrP species glycosylated at the first N-linked glycosylation site at residue 181 and they proposed that the deficiency in PrP glycosylation may be involved in the pathogenesis of the two conditions. ... BBC News. v t e Prion diseases and transmissible spongiform encephalopathy Prion diseases in humans inherited/ PRNP : fCJD Gerstmann–Sträussler–Scheinker syndrome Fatal familial insomnia sporadic: sCJD Sporadic fatal insomnia Variably protease-sensitive prionopathy acquired/ transmissible: iCJD vCJD Kuru Prion diseases in other animals Bovine spongiform encephalopathy Camel spongiform encephalopathy Scrapie Chronic wasting disease Transmissible mink encephalopathy Feline spongiform encephalopathy Exotic ungulate encephalopathy
Description IMD23 is an autosomal recessive primary immunodeficiency syndrome characterized by recurrent respiratory and skin infections beginning in early childhood. ... Hay et al. (2004) proposed the designation 'immunodeficiency-vasculitis-myoclonus syndrome' (IVMS). Zhang et al. (2014) presented a follow-up of the family reported by Hay et al. (2004) and also reported 3 affected males from a consanguineous Egyptian family.
PGM3-CDG is a rare congenital disorder of glycosylation caused by mutations in the PGM3 gene and characterized by neonatal to childhood onset of recurrent bacterial and viral infections, inflammatory skin diseases, atopic dermatitis and atopic diatheses, and marked serum IgE elevation. Early neurologic impairment is evident including developmental delay, intellectual disability, ataxia, dysarthria, sensorineural hearing loss, myoclonus and seizures.
PGM3 -congenital disorder of glycosylation ( PGM3 -CDG) is an inherited condition that primarily affects the immune system but can also involve other areas of the body. The pattern and severity of this disorder's signs and symptoms typically vary. Most people with PGM3 -CDG have impaired immune function (immune deficiency). Many have a shortage of white blood cells (leukopenia), which normally protect the body from infection. Because affected individuals lack the necessary immune cells to fight off certain bacteria, viruses, and fungi, they are prone to repeated and persistent infections that often occur in the lungs, ears, skin, or gastrointestinal tract.
Often patients present with similar manifestations to Hyperimmunoglobulin E syndrome (HIES), including severe atopic dermatitis , chronic sinusitis or otitis, cutaneous vasculitis , severe pulmonary infections and pneumonia , and very high concentrations of the serum antibody IgE levels. ... Recessive refers to each affected person needs two copies of the abnormal gene—one copy from each parent—to develop the syndrome. Typically, both parents of an affected child carry one abnormal gene and are unaffected by the disease.
Dermatophagia Extreme nail biting / biting of skin to point of an obsessive compulsive disorder (OCD) or other condition leading to self mutilating behavior such as autistic spectrum disorders (as is the case in this example) or Lesch-Nyhan Syndrome [ citation needed ] Specialty Psychiatry Types OCD A person with dermatophagia's extremely bitten finger The fingers of a person with dermatophagia. ... See also [ edit ] Excoriation disorder Lesch-Nyhan Syndrome Body-focused repetitive behavior Notes [ edit ] ^ Scott, MJ (January 1997).
The fracture may also cause damage to the arteries in the neck, resulting in lateral medullary syndrome , Horner's syndrome , ataxia , and the inability to sense pain or temperature. [1] In rare cases, congenital abnormality may cause the same symptoms as a Jefferson fracture. [3] [4] Cause [ edit ] Axial CT scan showing a Jefferson fracture.
A month later, the Chinese authorities claimed they were victims of Haff disease. [5] An outbreak was reported in Brooklyn, New York on 18 November 2011, when two household members were stricken by the syndrome after eating buffalo fish . [6] On February 4, 2014 two cases of Haff disease were reported in Cook County, Illinois following the consumption of buffalo fish. [7] A group from Brazil identified a Haff disease outbreak in the State of Bahia that lasted from December 2016 to April 2017, [8] with 67 cases identified. ... Jg.) pp. 57–60 External links [ edit ] Classification D Classification D ICD - 9-CM : 985.1 DiseasesDB : 33568 v t e Poisoning Toxicity Overdose History of poison Inorganic Metals Toxic metals Beryllium Cadmium Lead Mercury Nickel Silver Thallium Tin Dietary minerals Chromium Cobalt Copper Iron Manganese Zinc Metalloids Arsenic Nonmetals Sulfuric acid Selenium Chlorine Fluoride Organic Phosphorus Pesticides Aluminium phosphide Organophosphates Nitrogen Cyanide Nicotine Nitrogen dioxide poisoning CHO alcohol Ethanol Ethylene glycol Methanol Carbon monoxide Oxygen Toluene Pharmaceutical Drug overdoses Nervous Anticholinesterase Aspirin Barbiturates Benzodiazepines Cocaine Lithium Opioids Paracetamol Tricyclic antidepressants Cardiovascular Digoxin Dipyridamole Vitamin poisoning Vitamin A Vitamin D Vitamin E Megavitamin-B 6 syndrome Biological 1 Fish / seafood Ciguatera Haff disease Ichthyoallyeinotoxism Scombroid Shellfish poisoning Amnesic Diarrhetic Neurotoxic Paralytic Other vertebrates amphibian venom Batrachotoxin Bombesin Bufotenin Physalaemin birds / quail Coturnism snake venom Alpha-Bungarotoxin Ancrod Batroxobin Arthropods Arthropod bites and stings bee sting / bee venom Apamin Melittin scorpion venom Charybdotoxin spider venom Latrotoxin / Latrodectism Loxoscelism tick paralysis Plants / fungi Cinchonism Ergotism Lathyrism Locoism Mushrooms Strychnine 1 including venoms , toxins , foodborne illnesses .
"Polioencephalitis and the brain Fatigue Generator Model of Post-Viral Fatigue Syndromes". J. Of Chronic Fatigue Syndrome . 2 (2–3): 5–27. doi : 10.1300/J092v02n02_02 . ^ a b Racaniello VR (2006).
PCVD is caused by Porcine circovirus 2 (PCV-2). [3] The North American industry [4] [5] endorses "PCVAD" and European use "PCVD" to describe this disease. [6] [7] Contents 1 PMWS and PCV-2 2 Clinical signs 3 Management practices to decrease severity of PMWS 4 See also 5 References 6 Further reading PMWS and PCV-2 [ edit ] Postweaning multisystemic wasting syndrome ( PMWS ) is the classic PCVD entity, caused by PCV-2. [1] PCV-2 has a near universal distribution – present in most pig herds. ... Clinical signs [ edit ] Both PMWS and porcine dermatitis and nephropathy syndrome ( PDNS ) are associated to PCV-2. [8] Many pigs affected by the circovirus also seem to develop secondary bacterial infections, like Glässer disease ( Haemophilus parasuis ), pulmonary pasteurellosis, colibacilosis, salmonellosis and others. [9] Postmortem lesions occur in multiple organs, especially in lymphoid tissues and lung, giving rise to the term "multisystemic". [1] [2] [10] Lesions may also affect the skin, kidney, reproductive tissue, brain, or blood vessels. [1] Wasting pigs is the most common sign of PMWS infection, increasing the mortality rate significantly.
Aripiprazole which is known to have several very serious side effects may be useful for treatment of apathy syndrome (avolition). However, its role and efficacy in treatment of apathy requires further investigation in clinical trials. [7] Mitragynine contained in Kratom has the ability to reduce avolition. [8] The dopaminergic neurons of the prefrontal cortex are significantly reduced, and is associated with avolition. ... See also [ edit ] Abulia ADHD Akrasia Amotivational syndrome Anhedonia Apathy Incontinence Lethargy Volition (psychology) Hikikomori Learned helplessness Kratom References [ edit ] ^ American Psychiatric Association (2013).
Other neurological conditions Perseveration is a common feature of frontal lobe syndrome , as well as neurodegenerative diseases such as progressive supranuclear palsy , corticobasal syndrome and chronic acetogenin poisoning. [8] Perseveration may also refer to the obsessive and highly selective interests of individuals on the autism spectrum .
See also chromosome 3p deletion syndrome (613792). Clinical Features Yan et al. (2017) reported 10 patients from 9 unrelated families with global developmental delay apparent since infancy, mild to severe intellectual disability, expressive language impairment, and dysmorphic facial features. ... Mattioli et al. (2017) reported a large 3-generation family in which 6 individuals had an autosomal dominant syndromic form of mild intellectual disability associated with additional features such as growth retardation, ptosis, and relative microcephaly.
Some patients develop cardiac arrhythmias reminiscent of sick sinus syndrome (summary by Lodder et al., 2016 and Shamseldin et al., 2016). ... INHERITANCE - Autosomal recessive CARDIOVASCULAR Heart - Sick sinus syndrome (in some patients) - Bradycardia (in some patients) - Arrhythmias (in some patients) MUSCLE, SOFT TISSUES - Hypotonia (in some patients) NEUROLOGIC Central Nervous System - Developmental delay (in some patients) - Intellectual disability (in some patients) - Speech delay - Impaired fine motor skills (in some patients) Behavioral Psychiatric Manifestations - Attention deficit - Hyperactivity MISCELLANEOUS - Onset in early childhood - Three unrelated families have been reported (last curated November 2016) MOLECULAR BASIS - Caused by mutation in the guanine nucleotide-binding protein, beta-5 gene (GNB5, 604447.0006 ) ▲ Close