-
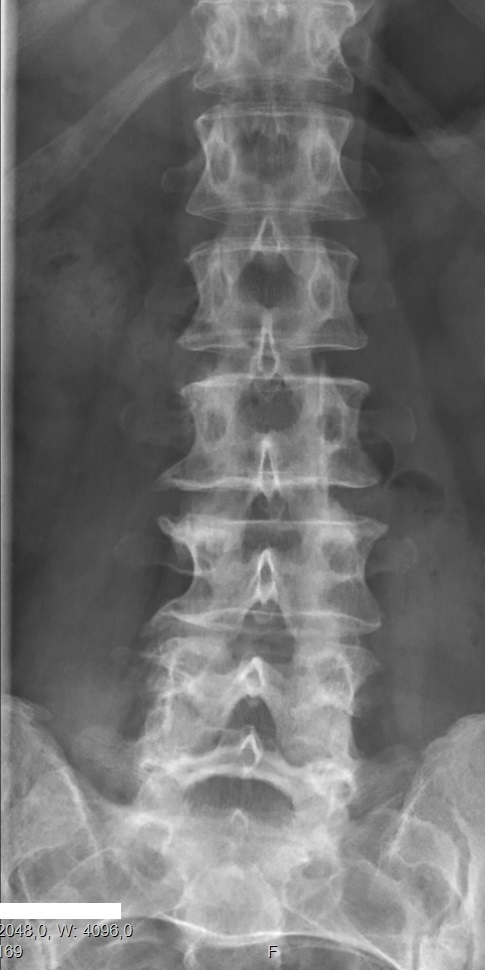
Bertolotti's Syndrome
Wikipedia
Bertolotti's syndrome Lumbarization of S1 Bertolotti's syndrome is a commonly missed cause of back pain which occurs due to lumbosacral transitional vertebrae (LSTV). ... "A Review of Symptomatic Lumbosacral Transitional Vertebrae: Bertolotti's Syndrome" . International Journal of Spine Surgery . 9 : 42. doi : 10.14444/2042 . ... "A Review of Symptomatic Lumbosacral Transitional Vertebrae: Bertolotti's Syndrome" . International Journal of Spine Surgery . 9 : 42. doi : 10.14444/2042 . ... "Lumbosacral transitional vertebra causing Bertolotti's syndrome: A case report and review of the literature" . ... External links [ edit ] Classification D ICD - 10 : Q76.49 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum
-
Haddad Syndrome
Orphanet
Haddad syndrome is a rare congenital disorder in which congenital central hypoventilation syndrome (CCHS), or Ondine syndrome, occurs concurrently with Hirschsprung disease (see these terms). Epidemiology Birth incidence of Ondine syndrome is 1 in 200,000 live-births and Hirschsprung disease occurs concurrently in 16% of cases. ... Etiology Mutations in the PHOX2B gene are found in a significant number of patients with Haddad syndrome.

-
Summitt Syndrome
Omim
The skull was towered, as in Carpenter syndrome (201000). The parents were first cousins. ... Cohen et al. (1987) concluded that Summitt syndrome and Goodman syndrome (201020) are variants of Carpenter syndrome. Head - Craniosynostosis Growth - Obesity Neuro - Normal intelligence - Tower skull Limbs - Syndactyly Inheritance - Autosomal recessive - ? variant of Carpenter syndrome ▲ Close
-
Hunter-Mcalpine Syndrome
Gard
Hunter-McAlpine syndrome is a very rare condition characterized by developmental delay, intellectual disability, and small head size (microcephaly). ... This can cause a misshapen skull and is common in individuals with Hunter-McAlpine syndrome; in fact another name for the condition is Hunter-McAlpine craniosynostosis syndrome. Hunter-McAlpine syndrome is a genetic condition, meaning that it is caused by changes in the genes. Hunter-McAlpine syndrome is typically diagnosed by genetic testing, and treatment options are focused on managing each individual’s symptoms.
-
Chandler's Syndrome
Gard
Chandler's syndrome is a rare eye disorder in which the single layer of cells lining the interior of the cornea proliferates, causing changes within the iris, corneal swelling, and unusually high pressure in the eye (glaucoma). This condition is one of three syndromes, along with progressive iris atrophy and Cogan-Reese syndrome, that make up the iridocorneal endothelial (ICE) syndrome . ... Symptoms may include reduced vision and pain. Chandler's syndrome more often affects females and usually presents sometime during middle age.
-
Terminal Osseous Dysplasia With Pigmentary Defects
Wikipedia
Terminal osseous dysplasia with pigmentary defects Specialty Dermatology Terminal osseous dysplasia with pigmentary defects is a cutaneous condition characterized by hyperpigmented, atrophic facial macules . [1] It has been associated with FLNA . [2] See also [ edit ] Corneodermatosseous syndrome Osseous choristoma of the tongue List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... External links [ edit ] Classification D OMIM : 300244 MeSH : C564554 v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins This dermatology article is a stub .
-
Multiple Familial Trichoepithelioma
Wikipedia
Multiple familial trichoepithelioma Other names Brooke–Spiegler syndrome and Epithelioma adenoides cysticum Specialty Dermatology Multiple familial trichoepithelioma is a cutaneous condition characterized by multiple cystic and solid nodules appearing on the face. [1] : 672 Contents 1 Classification 2 Brooke–Spiegler syndrome 3 See also 4 References Classification [ edit ] The classification of this syndrome is difficult. Three conditions are known to be caused by mutations in the CYLD gene: Brooke–Spiegler syndrome, multiple familial trichoepithelioma, and familial cylindromatosis . ... Types include: Type OMIM Gene Locus MFT1 601606 CYLD 16q12-q13 MFT2 612099 ? 9p21 Brooke–Spiegler syndrome [ edit ] Brooke–Spiegler syndrome (named after the dermatologists Henry Ambrose Grundy Brooke and Eduard Spiegler ) [2] is a condition in which multiple skin tumors develop from skin structures. Tumors commonly occurring in this syndrome include spiradenomas , trichoepitheliomas , and cylindromas . ... Philadelphia, PA: Elsevier. ^ http://omim.org/entry/605041 v t e Cancers of skin and associated structures Glands Sweat gland Eccrine Papillary eccrine adenoma Eccrine carcinoma Eccrine nevus Syringofibroadenoma Spiradenoma Apocrine Cylindroma Dermal cylindroma Syringocystadenoma papilliferum Papillary hidradenoma Hidrocystoma Apocrine gland carcinoma Apocrine nevus Eccrine / apocrine Syringoma Hidradenoma or Acrospiroma / Hidradenocarcinoma Ceruminous adenoma Sebaceous gland Nevus sebaceous Muir–Torre syndrome Sebaceous carcinoma Sebaceous adenoma Sebaceoma Sebaceous nevus syndrome Sebaceous hyperplasia Mantleoma Hair Pilomatricoma / Malignant pilomatricoma Trichoepithelioma Multiple familial trichoepithelioma Solitary trichoepithelioma Desmoplastic trichoepithelioma Generalized trichoepithelioma Trichodiscoma Trichoblastoma Fibrofolliculoma Trichilemmoma Trichilemmal carcinoma Proliferating trichilemmal cyst Giant solitary trichoepithelioma Trichoadenoma Trichofolliculoma Dilated pore Isthmicoma Fibrofolliculoma Perifollicular fibroma Birt–Hogg–Dubé syndrome Hamartoma Basaloid follicular hamartoma Folliculosebaceous cystic hamartoma Folliculosebaceous-apocrine hamartoma Nails Neoplasms of the nailbed v t e Disorders of translation and posttranslational modification Translation Ribosome : Diamond–Blackfan anemia FMR1 Fragile X syndrome Fragile X-associated tremor/ataxia syndrome Premature ovarian failure 1 Initiation factor : Leukoencephalopathy with vanishing white matter snRNP : Retinitis pigmentosa 33 Posttranslational modification Protein folding Alzheimer's disease Huntington's disease Creutzfeldt–Jakob disease chaperonins: 3-Methylglutaconic aciduria 5 Protein targeting I-cell disease Ubiquitin E1 : X-linked spinal muscular atrophy 2 E3 : Johanson–Blizzard syndrome Von Hippel–Lindau disease 3-M syndrome Angelman syndrome Deubiquitinating enzyme : Machado–Joseph disease Aneurysmal bone cyst Multiple familial trichoepithelioma 1 SUMO OFC10 Other Multiple sulfatase deficiency Hyperproinsulinemia Ehlers–Danlos syndrome 6CYLD, ERBB2, TP53, CDKN2A, PTGS2, COX2, MTCO2P12, ARID1A, APC, SMAD4, PIK3CA, TNF, AXL, CCND1, CDK4, ESR1, RUNX3, WIF1, PIK3CB, ADIPOQ, EGFR, MAPK1, PIK3CD, PIK3CG, EGF, MET, VEGFA, MIR31, AKT1, VDR, MDM2, LEP, CCN1, WNT3A, IGFBP3, EPHB2, MGMT, AFAP1-AS1, JUN, IL6, CXCL8, ITK, IGFBP2, MYC, IGF1, EPC1, MTHFR, MCL1, FGFR2, FOXF1, SUB1, GLI1, GSN, GSTP1, MMP1, HGD, EPC2, HSP90B1, BCL2, BECN1, HOMER2, CDK6, C9, CDK9, MIR203A, SLC22A3, MIR192, MIR148A, RUNX1, CTNNB1, XIAP, PARP1, MIR145, APEX1, FAM215A, TNNT1, TLR4, SMUG1, ADAM12, RNF19A, SLC35A2, DKK1, POLDIP2, TIMP3, UGT1A, TIMP1, XRCC1, TFF1, AIMP2, NOC2L, BTG3, HMGA2, RPP14, GRAP2, COX5A, KLF4, MELK, ELMO1, GAB2, EIF1, CDK2AP2, CLDN2, PPIE, HSPB3, ALDH1A2, OLFM4, IL18RAP, MRPL28, AHSA1, TXNIP, ADAM9, TRIM31, AKAP12, ADAM10, RABGEF1, MIR15B, MIR21, MIR205, MIR200A, MIR191, MIR187, MIR17, MIR130A, MIR217, MIR106B, MIR106A, C6orf120, LCE1E, HNF1A-AS1, SEC14L3, MIR210, MIR23B, METTL7B, MIR505, H3P8, H3P9, COMMD3-BMI1, HPP1, MIR652, FABP12, MIR202, MIR27B, MIR378A, MIR375, MIR331, MIR133B, MIR7-2, MIR30A, JMJD1C, GDF7, LAMTOR2, CRNKL1, TERF2IP, TBX5, WWOX, GHRL, SF3B6, VTA1, TNFRSF12A, BNC2, PLCE1, INSIG2, RNF141, UHRF1, CD274, NXT1, RIN2, BARX1, GPBAR1, MAP1LC3B, IL23R, FOXR2, CTHRC1, OMA1, SLC52A3, MTDH, MUS81, AKR1B10, C12orf49, RNF128, EPS8L3, VSIR, SRR, GJC2, TERT, PTGS1, TAP2, CTSE, CYP17A1, DAD1, CD55, DMD, ATN1, DUSP6, E2F3, MEGF8, EIF4A2, EIF4E, EIF4G1, ELANE, EMX2, ENG, ERBB3, ERCC1, ERCC2, ERCC4, ESR2, FABP1, FABP6, FBLN1, FGFR3, FHIT, FKBP4, FKBP5, MTOR, FUT4, GHR, CX3CR1, CTSB, GSTT2, MAPK14, AGT, AHR, AMCN, ANXA1, APOB, AR, ATM, BMI1, BMP4, BRCA1, BRCA2, CA2, CA9, CA12, CAMK2G, CASP3, CASP7, CCKBR, CD80, CD34, CDH1, CDH13, CDKN2B, CDX2, COL1A1, COL11A2, COX8A, CPOX, CRK, GPX2, HLA-B, TAC1, PECAM1, PGR, PKP1, PPARG, PRKDC, MAPK8, KLK7, PTEN, PTPRC, PVT1, RAC1, RAP1A, RAP1GAP, RBM3, RFC3, RORC, RPS6KB1, RRAD, S100A9, SAFB, SERPINB3, SERPINB4, SLC10A2, SMO, SOX2, SOX4, SOX9, SPP1, STAT3, STK11, SERPINF1, SLC22A18, HLA-C, NELL1, HNF4A, HSPA4, HSPB1, HSPB2, HTC2, IFNG, IGF1R, IGF2, IL1A, IL1B, IL17A, IL18, IDO1, INHBA, LGALS9, LTA, EPCAM, MAGEA6, MCM5, MLH1, MMP3, MPG, MPO, MSH2, MSI1, MSR1, COX1, MUC6, MYO9B, H3P10
-
Saul-Wilson Syndrome
Medlineplus
Saul-Wilson syndrome is characterized by short stature (dwarfism) and other skeletal abnormalities. ... In Saul-Wilson syndrome, levels of white blood cells can vary from normal to low (intermittent neutropenia). Neutropenia makes it more difficult for the body to fight off foreign invaders such as bacteria and viruses, and may contribute to recurrent respiratory infections that occur in childhood in some individuals with Saul-Wilson syndrome. Frequency Saul-Wilson syndrome is a very rare disorder. At least 16 affected individuals have been reported in the scientific literature. Causes Saul-Wilson syndrome is caused by mutations in the COG4 gene. ... The COG4 gene mutations that cause Saul-Wilson syndrome result in production of an abnormal COG4 protein.
-
Alport Syndrome
Medlineplus
Alport syndrome is a genetic condition characterized by kidney disease, hearing loss, and eye abnormalities. People with Alport syndrome experience progressive loss of kidney function. ... Significant hearing loss, eye abnormalities, and progressive kidney disease are more common in males with Alport syndrome than in affected females. Frequency Alport syndrome occurs in approximately 1 in 50,000 newborns. ... Learn more about the genes associated with Alport syndrome COL4A3 COL4A4 COL4A5 Inheritance Pattern Alport syndrome can have different inheritance patterns. ... People with this form of Alport syndrome have one mutation in either the COL4A3 or COL4A4 gene in each cell.
-
Crest Syndrome
Wikipedia
Unsourced material may be challenged and removed. Find sources: "CREST syndrome" – news · newspapers · books · scholar · JSTOR ( February 2009 ) ( Learn how and when to remove this template message ) CREST syndrome Other names Calcinosis-Raynaud phenomenon-esophageal involvement-sclerodactyly-telangiectasia syndrome [1] CREST syndrome (calcinosis and sclerodactyly) Specialty Rheumatology CREST syndrome , also known as the limited cutaneous form of systemic sclerosis (lcSSc), is a multisystem connective tissue disorder . ... Contents 1 Signs and symptoms 1.1 Calcinosis 1.2 Raynaud's phenomenon 1.3 Esophageal dysmotility 1.4 Sclerodactyly 1.5 Telangiectasias 1.6 Other 2 Cause 3 Diagnosis 4 Treatment 5 Epidemiology 5.1 History 6 See also 7 References 8 External links Signs and symptoms [ edit ] CREST syndrome (calcinosis and sclerodactyly) X-rays showing calcinosis in a woman with CREST syndrome. ... "Multiple telangiectasia, Raynaud's phenomenon, sclerodactyly, and subcutanious calcinosis: a syndrome mimicking hereditary hemorrhagic telangiectasia". ... Retrieved 2014-02-20 . ^ "CREST syndrome: MedlinePlus Medical Encyclopedia Image" . medlineplus.gov . ^ "CREST syndrome - Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ^ Talwalkar, JA; Lindor, KD (Jul 5, 2003). ... External links [ edit ] Classification D ICD - 10 : M34.1 ICD - 9-CM : 710.1 OMIM : 181750 MeSH : D017675 DiseasesDB : 29764 v t e Systemic connective tissue disorders General Systemic lupus erythematosus Drug-induced SLE Libman–Sacks endocarditis Inflammatory myopathy Myositis Dermatopolymyositis Dermatomyositis / Juvenile dermatomyositis Polymyositis * Inclusion body myositis Scleroderma Systemic scleroderma Progressive systemic sclerosis CREST syndrome Overlap syndrome / Mixed connective tissue disease Other hypersensitivity / autoimmune Sjögren syndrome Other Behçet's disease Polymyalgia rheumatica Eosinophilic fasciitis Eosinophilia–myalgia syndrome fibrillin Marfan syndrome Congenital contractural arachnodactylyHLA-DRB1, KIAA0319L, CAV1, IRF5, CCN2, CCR6, SOX10, IL6, FAS, CD22, TAP1, MITF, MRC1, NOS1, NOS2, PAH, CCL4, CCL18, SOX5, ACVRL1, SSB, TAP2, TGFB1, TIMP1, TNF, HSP90B2P, TTR, VIM, RABEP1, SS18L1, IL21R, RXFP1, JAK2, CXCL10, CD86, GRB10, CHI3L1, CCR1, C1QC, C1QB, CTSV, GADD45A, EGFR, FCGR3A, FCGR3B, FYN, HLA-B, IL13, CCND1, HMGB1, HOXC8, DNAJB1, IRF8, IFI16, IL2, ACTB, CXCL8, IL10, ACR

-
Primrose Syndrome
Wikipedia
Primrose syndrome Other names Intellectual disability-cataracts-calcified pinnae-myopathy syndrome Primrose syndrome is inherited via an autosomal dominant manner [1] Primrose syndrome is a rare, slowly progressive genetic disorder that can vary symptomatically between individual cases, but is generally characterised by ossification of the external ears , learning difficulties , and facial abnormalities. [2] It was first described in 1982 in Scotland 's Royal National Larbert Institution by Dr D.A.A. Primrose. [3] Primrose syndrome appears to occur spontaneously, regardless of family history. ... Whether or not this deletion is related to the syndrome or is a harmless mutation is unknown. ... PMID 20644156 . ^ a b "Primrose syndrome" . Office of Rare Diseases Research . ... External links [ edit ] Primrose syndrome at Online Mendelian Inheritance in Man Primrose syndrome at The National Library of Medicine archives Journal of Neurology article Classification D ICD - 10 : Q87.8 OMIM : 259050 MeSH : C536420 External resources Orphanet : 3042

-
Behr Syndrome
Wikipedia
Behr syndrome Other names Optic atrophy in early childhood, associated with ataxia, spasticity, mental retardation, and posterior column sensory loss Behr syndrome has an autosomal recessive pattern of inheritance . ... P. (2008-01-01). "Familial Behr syndrome-like phenotype with autosomal dominant inheritance". ... "Heterozygous OPA1 mutations in Behr syndrome" . Brain . 134 (Pt 4): e169, author reply e170. doi : 10.1093/brain/awq306 . ... "Musculoskeletal deformities in Behr syndrome". Journal of Pediatric Orthopedics . 21 (4): 512–514. doi : 10.1097/01241398-200107000-00018 . ... PMID 8060430 . External links [ edit ] Behr syndrome at NIH 's Office of Rare Diseases Behr Syndrome at Omim.com Classification D ICD - 10 : G11.18 OMIM : 210000 MeSH : C537669 DiseasesDB : 32611

-
Klüver–bucy Syndrome
Wikipedia
Syndrome resulting from bilateral lesions of the medial temporal lobe Klüver-Bucy syndrome Specialty Psychiatry , neurology Klüver–Bucy syndrome is a syndrome resulting from bilateral lesions of the medial temporal lobe (including amygdaloid nucleus). [1] Klüver–Bucy syndrome may present with compulsive eating , hypersexuality , insertion of inappropriate objects in the mouth (hyperorality), visual agnosia , and docility . ... ISBN 978-0-300-05271-8 . Klüver-Bucy syndrome. ^ Terzian, H.; Ore, G.D. (1955). "Syndrome of Klüver and Bucy; reproduced in man by bilateral removal of the temporal lobes" . ... ISBN 978-0-521-86001-7 . Klüver-Bucy syndrome. ^ Marlowe WB, Mancall EL, Thomas JJ (1975). "Complete Klüver-Bucy syndrome in man". Cortex . 11 (1): 53–9. doi : 10.1016/s0010-9452(75)80020-7 .
-
Proximal 18q Deletion Syndrome
Medlineplus
Proximal 18q deletion syndrome is a chromosomal condition that occurs when a piece of the long (q) arm of chromosome 18 is missing. ... Individuals with proximal 18q deletion syndrome have a wide variety of signs and symptoms. ... However, only a small number of these individuals have deletions in the region associated with proximal 18q deletion syndrome. At least 15 people with proximal 18q deletion syndrome have been described in the medical literature. Causes Proximal 18q deletion syndrome is caused by a deletion of genetic material from one copy of chromosome 18. ... Learn more about the chromosome associated with Proximal 18q deletion syndrome chromosome 18 Inheritance Pattern Proximal 18q deletion syndrome is considered to be an autosomal dominant condition.

-
Leukotriene Receptor Antagonist-Associated Churg–strauss Syndrome
Wikipedia
Leukotriene receptor antagonist-associated Churg–Strauss syndrome Leukotriene receptor antagonist-associated Churg–Strauss syndrome may occur in asthma patients being treated with leukotriene receptor antagonists , occurring 2 days to 10 months after the antagonist has been started, with features of the syndrome including peripheral eosinophilia , pulmonary infiltrates , and less commonly neuropathy , sinusitis , and cardiomyopathy . [1] : 135–6 See also [ edit ] Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) Leukotriene antagonist Skin lesion References [ edit ] ^ James, William; Berger, Timothy; Elston, Dirk (2005). ... External links [ edit ] Classification D ICD - 10 : Y55.6 ICD - 9-CM : E945.7 v t e Adverse drug reactions Antibiotics Penicillin drug reaction Sulfonamide hypersensitivity syndrome Urticarial erythema multiforme Adverse effects of fluoroquinolones Red man syndrome Jarisch–Herxheimer reaction Hormones Steroid acne Steroid folliculitis Chemotherapy Chemotherapy-induced acral erythema Chemotherapy-induced hyperpigmentation Scleroderma-like reaction to taxanes Hydroxyurea dermopathy Exudative hyponychial dermatitis Anticoagulants Anticoagulant-induced skin necrosis Warfarin necrosis Vitamin K reaction Texier's disease Immunologics Adverse reaction to biologic agents Leukotriene receptor antagonist-associated Churg–Strauss syndrome Methotrexate-induced papular eruption Adverse reaction to cytokines Other drugs Anticonvulsant hypersensitivity syndrome Allopurinol hypersensitivity syndrome Vaccine adverse event Eczema vaccinatum Bromoderma Halogenoderma Iododerma General Skin and body membranes Acute generalized exanthematous pustulosis Bullous drug reaction Drug-induced acne Drug-induced angioedema Drug-related gingival hyperplasia Drug-induced lichenoid reaction Drug-induced lupus erythematosus Drug-induced nail changes Drug-induced pigmentation Drug-induced urticaria Stevens–Johnson syndrome Injection site reaction Linear IgA bullous dermatosis Toxic epidermal necrolysis HIV disease-related drug reaction Photosensitive drug reaction Other Drug-induced pseudolymphoma Fixed drug reaction Serum sickness-like reaction This cutaneous condition article is a stub .
-
Trigeminal Trophic Syndrome
Wikipedia
Trigeminal trophic syndrome Other names Trigeminal trophic lesion Trigeminal trophic syndrome is a rare disease caused by the interruption of peripheral or central sensory pathways of the trigeminal nerve . ... (May 2004). "Trigeminal trophic syndrome--report of four cases and review of the literature". ... PMID 15099331 . v t e Neurotrauma Traumatic brain injury Intracranial hemorrhage Intra-axial Intraparenchymal hemorrhage Intraventricular hemorrhage Extra-axial Subdural hematoma Epidural hematoma Subarachnoid hemorrhage Brain herniation Cerebral contusion Cerebral laceration Concussion Post-concussion syndrome Second-impact syndrome Dementia pugilistica Chronic traumatic encephalopathy Diffuse axonal injury Abusive head trauma Penetrating head injury Spinal cord injury Anterior spinal artery syndrome Brown-Séquard syndrome Cauda equina syndrome Central cord syndrome Paraplegia Posterior cord syndrome Spinal cord injury without radiographic abnormality Tetraplegia (Quadriplegia) Peripheral nerves Nerve injury Peripheral nerve injury classification Wallerian degeneration Injury of accessory nerve Brachial plexus injury Traumatic neuroma This cutaneous condition article is a stub .

-
Quadrilateral Space Syndrome
Wikipedia
Quadrilateral space syndrome Other names Quadrangular space syndrome Shoulder muscle (rotator cuff) Specialty Neurology Quadrilateral space syndrome is a rotator cuff denervation syndrome in which the axillary nerve is compressed at the quadrilateral space of the rotator cuff. ... Atrophy can occur in cases of chronic nerve impingement. [1] [2] It can be associated with a glenoid labral cyst, with the cyst also reflecting injury of the glenoid labrum . [3] Differential diagnosis [ edit ] Differential considerations include similar rotator cuff denervation syndromes such as Parsonage–Turner syndrome , and compression of the suprascapular nerve at the spinoglenoid notch in which the infraspinatus , and to a lesser degree supraspinatus is involved. ... Lee; Helms, Clyde (2005-03-01). "Quadrilateral space syndrome: incidence of imaging findings in a population referred for MRI of the shoulder". ... "Ultrasound findings of teres minor denervation in suspected quadrilateral space syndrome". Journal of Clinical Ultrasound . 34 (7): 343–347. doi : 10.1002/jcu.20239 . ... S. (2000-10-01). "Quadrilateral space syndrome caused by glenoid labral cyst".

-
Wagner Syndrome
Medlineplus
Wagner syndrome is a hereditary disorder that causes progressive vision loss. ... Vision impairment in people with Wagner syndrome can vary from near normal vision to complete loss of vision in both eyes. Frequency Wagner syndrome is a rare disorder, although its exact prevalence is unknown. ... Causes Mutations in the VCAN gene cause Wagner syndrome. The VCAN gene provides instructions for making a protein called versican. ... VCAN gene mutations that cause Wagner syndrome lead to insufficient levels of versican in the vitreous.
-
Van Der Woude Syndrome
Medlineplus
Van der Woude syndrome is a condition that affects the development of the face. ... In some cases, people with van der Woude syndrome have missing teeth. People with van der Woude syndrome who have cleft lip and/or palate, like other individuals with these facial conditions, have an increased risk of delayed language development, learning disabilities, or other mild cognitive problems. The average IQ of individuals with van der Woude syndrome is not significantly different from that of the general population. Frequency Van der Woude syndrome is believed to occur in 1 in 35,000 to 1 in 100,000 people, based on data from Europe and Asia. ... Mutations in the IRF6 gene that cause van der Woude syndrome prevent one copy of the gene in each cell from making any functional protein.IRF6, GRHL3, TNF, IL6, IL1B, TLR4, IL10, ACTB, IL17A, IFNG, CXCL8, COX2, HMGB1, NHLH1, PPARG, PTGS2, STAT3, HSPA4, MTCO2P12, AKT1, CYBB, AIF1, AHR, DCDC2, NOX4, AGO2, SIRT1, TNFSF13, UMOD, TRPC3, TP53, ETV6, TH, TGFB2, TFDP1, TFAP2A, CNTN2, SYT1, ARG1, SLC22A4, CCL21, CCL5, RIPK4, GORASP1, ROCK1, WNK1, THRIL, NCF1, MIR211, MIR206, MIR195, MIR183, MIR17, MIR15B, MIR132, MIR122, MALAT1, CFAP57, TICAM1, TRIM69, CPO, TIRAP, NLRP3, WDR26, WNK2, RORA, REN, F3, DNTT, CCK, IRF3, IRF2, IDO1, CDH2, CNR2, CXCR1, IL7R, CYP2E1, DRD2, RELA, IFNA13, IFNA1, HSP90AA1, EPHB2, FOXA1, ERN1, CXCL2, GPT, GLI2, KISS1, LAMB2, MAP3K3, MST1, NECTIN1, PVR, CCND1, MAPK1, PRKAB1, PRKAA2, PRKAA1, BCL2, AP2A1, PIK3CG, PIK3CD, PIK3CB, PIK3CA, NRF1, NME2, NME1, CASP3, NFATC1, CAV1, PKM
-
Multiple Synostoses Syndrome 2
Omim
A number sign (#) is used with this entry because of evidence that multiple synostoses syndrome-2 (SYNS2) is caused by heterozygous mutation in the GDF5 gene (601146) on chromosome 20q11. ... These findings were considered consistent with the syndrome described by Pearlman (see 186400) but showed considerable overlap with other multiple synostosis syndromes. ... Mapping Akarsu et al. (1999) mapped the multiple synostoses syndrome in an Iranian family to markers on chromosome 20q11.2, with the highest lod score observed at D20S200 (Z = 13.58, theta = 0.0). ... Molecular Genetics By mutation screening of a proband with multiple synostoses syndrome, Akarsu et al. (1999) identified a heterozygous missense mutation in the GDF5 gene (S475N; 601146.0013). ... Unlike mutations that lead to haploinsufficiency for GDF5 and produce brachydactyly C (113100), the protein encoded by the multiple synostoses syndrome allele was secreted as a mature GDF5 dimer.







