Complications occur in about half of hospitalized patients and include disseminated intravascular coagulopathy and acute respiratory distress syndrome and less frequently liver, renal or congestive heart failure, coma and death.
Babesiosis refers to a condition caused by microscopic parasites that infect the red blood cells. Many people who are infected with Babesia parasites do not experience any symptoms of the condition. When present, signs and symptoms may include flu-like symptoms such as fever, chills, headache, body aches, nausea and fatigue. In severe cases, babesiosis can be associated with hemolytic anemia. Babesia parasites are primarily spread by infected ticks. Treatment is generally only required in people who develop symptoms of the condition.
Brain imaging, performed in 3 patients, showed T2-weighted hyperintensities variably apparent in the thalamus, basal ganglia, and/or brainstem, suggestive of Leigh syndrome (256000). The severity was variable, ranging from death from cardiac failure in early infancy to survival into the second decade with mild intellectual disability.
A rare mitochondrial disease characterized by early onset of hypertrophic cardiomyopathy and variable neurologic symptoms including global developmental delay, hypotonia, intellectual disability, visual impairment, and seizures. Lactic acidosis is present in all patients. Muscle biopsy usually shows decreased activity of mitochondrial complexes I and IV. Brain imaging may reveal variable abnormal signal intensities in the thalamus, basal ganglia, and/or brain stem.
The main types of extra-axial hemorrhage are epidural hematoma (bleeding between the dura mater and the skull), subdural hematoma (in the subdural space ) and subarachnoid hemorrhage (between the arachnoid mater and pia mater ). Most of the hemorrhagic stroke syndromes have specific symptoms (e.g., headache , previous head injury ). [ citation needed ] Symptoms and signs [ edit ] Symptoms often include: [ citation needed ] Seizures, especially in newborns Keeping one hand in a fist position, especially in infants Worsening or sudden headaches Sudden difficulty speaking, slurring of words or trouble understanding speech Hemiparesis, or a weakness on one side of the body Sudden loss of vision or abnormal eye movements Sudden loss of balance or trouble walking Prognosis [ edit ] The prognosis for pediatric stroke survivors varies.
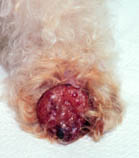
Malakoplakia is a chronic multisystem granulomatous inflammatory disease characterized by the presence of single or multiple soft plaques on various organs of the body. Epidemiology Prevalence is unknown but more than 700 cases have been described in the literature. Clinical description Malakoplakia can occur in all ages, with a mean age at diagnosis of 50 years old and a female predominance. Pediatric cases are rare. It is most common in immunodeficient patients with a history of diabetes, transplantation, lymphoma, steroid therapy or alcoholism. In the majority of cases (60-80%), malakoplakia affects the urinary tract (bladder, kidney, and ureters) and cases of locoregional extension have been reported (retroperitoneal region and lymph nodes).
Malakoplakia is a rare chronic inflammatory disease. It most commonly involves the urogenital system (reproductive organs and urinary system), but may also be found in other regions of the body, including the pelvis, bones, lungs, thyroid gland , gastrointestinal (digestive) tract, skin, and kidneys. Symptoms of malakoplakia differ depending on the involved area. For example, when the skin is affected the malakoplakia may appear rash-like with small areas of itchy, reddened skin that may be painful and/or fluid filled. The cause of malakoplakia is not well understood. It is thought to be related to an issue with the function of one's macrophages, which are one type of cell within the immune system that respond to foreign invaders (bacteria). E.coli is the most common type of bacteria that leads to malakoplakia. Individuals with a compromised immune system have an elevated risk to develop malakoplakia.
Spanheimer et al. (1982) reported 3 cases, the youngest a 4-year-old girl with goiter and symptoms of hyperthyroidism. The syndrome was attributed to selective pituitary insensitivity to thyroid hormone.
A rare genetic hyperthyroidism characterized by elevated levels of circulating free thyroid hormones, normal or elevated thyroid-stimulating hormone, decreased peripheral tissue responses to iodothyronine action, and a highly variable clinical phenotype which most commonly includes goiter, resting tachycardia, osteoporosis, short stature, and attention deficit disorder. Some patients may be entirely asymptomatic.
One of the probands (see 600820.0003) was also heterozygous for a missense mutation in the SYT1 gene (D233N; see 185605), which Izumi et al. (2016) suggested might have contributed to the additional feature of seizures in that patient; however, given that all 4 patients with ARCN1 mutations exhibited comparable degrees of developmental delay and intellectual disability, the authors concluded that the ARCN1 mutations likely play a major role in the neurologic features observed in this syndrome, and that ARCN1 is probably required for normal brain growth and cognitive development.
Description Immunodeficiency-47 is an X-linked recessive complex immunodeficiency syndrome characterized by recurrent bacterial infections, hypogammaglobulinemia, liver dysfunction, and defective glycosylation of serum proteins.
Neurological features may include cerebellar dysfunction (ataxia, nystagmus, tremor), axonal or demyelinating peripheral neuropathy (sensory and/or motor disturbances), cognitive impairment (dysexecutive syndrome, dementia), sensory impairment (optic, auditive neuropathy), epilepsy, myopathic features (ptosis, opthalmoparesis), extrapyramidal features (Parkinsonism, chorea, dystonia), psychiatric disturbances, and brain and spine imaging abnormalities (brain white matter alterations, thin corpus callosum, brain iron accumulation, cerebellar atrophy) which may be suggestive of a genetic subtype.
Description Leber congenital amaurosis with early-onset deafness is an autosomal dominant syndrome manifesting as early-onset and severe photoreceptor and cochlear cell loss.
The study by Najmabadi et al. (2011) included homozygosity mapping followed by exon enrichment and next-generation sequencing in 136 consanguineous families (over 90% Iranian and less than 10% Turkish or Arabic) segregating syndromic or nonsyndromic forms of autosomal recessive intellectual disability.
National Cancer Institute document: "Dictionary of Cancer Terms" . v t e Skin cancer of nevi and melanomas Melanoma Mucosal melanoma Superficial spreading melanoma Nodular melanoma lentigo Lentigo maligna / Lentigo maligna melanoma Acral lentiginous melanoma Amelanotic melanoma Desmoplastic melanoma Melanoma with features of a Spitz nevus Melanoma with small nevus-like cells Polypoid melanoma Nevoid melanoma Melanocytic tumors of uncertain malignant potential Nevus / melanocytic nevus Nevus of Ito / Nevus of Ota Spitz nevus Pigmented spindle cell nevus Halo nevus Pseudomelanoma Blue nevus of Jadassohn–Tièche Cellular Epithelioid Deep penetrating Amelanotic Malignant Congenital melanocytic nevus ( Giant Medium-sized Small-sized ) Balloon cell nevus Dysplastic nevus / Dysplastic nevus syndrome Acral nevus Becker's nevus Benign melanocytic nevus Nevus spilus This cutaneous condition article is a stub .
One such family has been reported. Description The MCIDDS syndrome is characterized by microcephaly and growth retardation, congenital cataracts, impaired intellectual development with attention deficit-hyperactivity disorder, and dystonia, with striatal thinning seen on MRI (Al-Owain et al., 2013).
In particular, the tonic tensor tympani syndrome . [6] [7] In France, researchers report the study of a case of acoustic shock in a scientific publication.