Diagnosis Since 2010 a description of the characteristic symptoms and findings of epilepsy of infancy with migrating focal seizures (EIMFS) has been included in the classification of epilepsy syndromes by the International League Against Epilepsy. ... Epilepsy features Seizure onset before age six months Developmental delay or developmental regression with seizure onset Seizure type At onset in most children: focal motor seizures that also frequently involve head and eye deviation Multifocal seizures proving intractable to conventional antiepileptic drugs Epilepsy syndromes. Epilepsy of infancy with migrating focal seizures EEG findings Interictal multifocal spikes In a single seizure, ictal-independent, unilateral, and migrating involvement of varying cortical areas with clinical-EEG correlation Initial EEG may be normal shortly after seizure onset, but epileptiform abnormalities are usually present within one month after first presentation. ... Prevalence To date, nine probands with SLC12A5- related epilepsy have been reported. The clinical syndrome epilepsy of infancy with migrating focal seizures (EIMFS) of all causes is itself rare.
The precocious formation of bilirubinate gallstones is the most common complication of hereditary spherocytosis, and the prevention of this problem represents a major impetus for splenectomy in many patients with compensated hemolysis. Because Gilbert syndrome (143500) had been considered a risk factor for gallstone formation, Miraglia del Giudice et al. (1999) postulated that the association of this common inherited disorder of hepatic bilirubin metabolism with hereditary spherocytosis could increase cholelithiasis. ... The 2-bp TA insertion within the promoter of the UGT1A1 gene (191740.0011), which is associated with Gilbert syndrome, was screened. The risk of developing gallstones was statistically different among the 3 groups of patients (homozygotes for the normal UGT1A1 allele, heterozygotes, and homozygotes for the allele with the TA insertion). ... Mice deficient in ankyrin have, in addition to hemolytic anemia, significant neurologic dysfunction associated with Purkinje cell degeneration in the cerebellum and the development of a late-onset neurologic syndrome characterized by persistent tremor and gait disturbance (Peters et al., 1991).
A number sign (#) is used with this entry because spherocytosis type 2 (SPH2) is caused by heterozygous mutation in the SPTB gene (182870) on chromosome 14q23. Some patients have been reported with homozygous or compound heterozygous mutations. Description Hereditary spherocytosis refers to a group of heterogeneous disorders that are characterized by the presence of spherical-shaped erythrocytes (spherocytes) on the peripheral blood smear. The disorders are characterized clinically by anemia, jaundice, and splenomegaly, with variable severity. Common complications include cholelithiasis, hemolytic episodes, and aplastic crises (review by Perrotta et al., 2008).
A number sign (#) is used with this entry because spherocytosis type 4 (SPH4) is caused by heterozygous mutation in the band 3 gene (SLC4A1, EPB3; 109270) on chromosome 17q21. For a general description and a discussion of genetic heterogeneity of spherocytosis, see SPH1 (182900). Clinical Features Prchal et al. (1991) studied a family with autosomal dominant hereditary spherocytosis associated with deficiency of erythrocyte band 3 protein. Del Giudice et al. (1992) reported a family in which a dominantly inherited form of hereditary spherocytosis was associated with deficiency of band 3, resulting in an increased spectrin/band 3 ratio. Since deficiency of spectrin is a much more frequent cause of hereditary spherocytosis, the usual finding is a decreased spectrin/band 3 ratio.
A number sign (#) is used with this entry because hereditary spherocytosis-3 is caused by mutation in the alpha-spectrin gene (SPTA; 182860) on chromosome 1q21. For a general description and a discussion of heterogeneity of spherocytosis, see 182900. Clinical Features Agre et al. (1982) reported 2 daughters, of related but normal parents, who had nearly fatal hemolytic anemia requiring early splenectomy. Both improved strikingly thereafter but spherocytosis persisted. Red cell membranes were at least 50% deficient in spectrin, with band 1 reduced more than band 2. No defect was found in membrane binding of spectrin or in membrane binding sites (ankyrin).
A number sign (#) is used with this entry because hereditary spherocytosis type 5 is caused by mutation in the gene encoding protein 4.2 (EPB42; 177070) on chromosome 15q15. For a general phenotypic description and a discussion of genetic heterogeneity of hereditary spherocytosis, see SPH1 (182900). Clinical Features Hereditary spherocytosis type 5, which has been observed predominantly in Japanese individuals, is an autosomal recessive disorder that results in a hemolytic anemia associated with abnormally shaped, osmotically fragile red blood cells (Bouhassira et al., 1992). Hayashi et al. (1974) described 4 Japanese patients, 3 of whom were sibs, with hereditary spherocytosis and deficiency of protein 4.2. Nozawa et al. (1974) reported a severe case of hereditary spherocytosis in a 6-year-old Japanese girl with protein 4.2 deficiency who showed improvement with splenectomy.
Hereditary spherocytosis is a condition characterized by hemolytic anemia (when red blood cells are destroyed earlier than normal). Signs and symptoms can range from mild to severe and may include pale skin, fatigue, anemia, jaundice, gallstones , and/or enlargement of the spleen . Other symptoms of hemolytic anemia may include feeling that your heart is pounding or racing (palpitations), feeling dizzy, problems concentrating, and headaches. Some people with a severe form of hereditary spherocytosis may have short stature, delayed puberty, and skeletal abnormalities. The condition is caused by mutations in any of several genes, such as the ANK1 , EPB42 , SLC4A1 , SPTA1 , and SPTB genes.
Sarcopenia is considered a component of frailty syndrome . [1] Sarcopenia can lead to reduced quality of life and disability. ... In the elderly, this often leads to decreased biological reserve and increased vulnerability to stressors known as the " frailty syndrome ". Loss of lean body mass is also associated with increased risk of infection, decreased immunity, and poor wound healing. ... Identification and research on potential therapeutic approaches and timing of interventions is also needed. [33] New pharmaceutical therapies in clinical development include myostatin and the selective androgen receptor modulators (SARMs). [34] Nonsteriodal SARMs are of particular interest, given they exhibit significant selectivity between the anabolic effects of testosterone on muscle, but with little to no evidence of androgenic effects (such as prostate stimulation in men). [34] See also [ edit ] Cachexia Ageing Frailty syndrome Geriatrics References [ edit ] ^ a b c Peterson SJ, Mozer M (February 2017).
T-ALL is not contagious nor inherited but specific genetic mutations, commonly including NOTCH1 and CDKN2A , may be passed along which increases susceptibility of T-ALL. [10] Causes of T-ALL [ edit ] Genetic conditions [ edit ] Some patients may have familial histories with leukemia predispositions which increases risk of developing T-ALL. Li-Fraumeni syndrome is an inherited condition that leads to mutation of TP53 , a tumor suppressor gene , which then increases risk of T-ALL. ... Furthermore, hereditary conditions such as Down syndrome , neurofibromatosis type 1 , ataxia telangiectasia , and Noonan syndrome are associated with higher risk of developing T-ALL.
A rare acute lymphoblastic leukemia characterized by a neoplasm of lymphoblasts committed to the T-cell lineage, involving bone marrow and blood. A value of >25% bone marrow blasts may be used to define leukemia (as opposed to lymphoma) in cases with the presence of a mass lesion in addition to bone marrow involvement. Patients typically present with leukocytosis, and frequently with a large mediastinal or other tissue mass. Lymphadenopathy and hepatosplenomegaly are common.
Treatment with pregabalin leads to a reduction in pain intensity of 50% or more in one person out of every 4-5 people treated (NNT=4-5). [9] Similarly, treatment with gabapentin also leads to a 50% reduction in pain intensity in one person out of every 7-8 people treated (NNT=7.5). [9] Opioids such as tramadol , methadone , oxycodone , and morphine have not been well-studied for postherpetic neuralgia treatment. [10] [11] [12] [13] Acetaminophen and nonsteroidal anti-inflammatory drugs are thought to be ineffective and have not undergone rigorous study for PHN. [1] [14] Prognosis [ edit ] The natural history of postherpetic neuralgia involves slow resolution of the pain syndrome. A subgroup of affected individuals may develop severe, long-lasting pain that does not respond to medical therapy. [ citation needed ] Epidemiology [ edit ] In the United States each year approximately 1,000,000 individuals develop herpes zoster. [15] Of those individuals, approximately 10-18% develop postherpetic neuralgia. [16] Fewer than 10 percent of people younger than 60 develop postherpetic neuralgia after a bout of herpes zoster, while about 40 percent of people older than 60 do. [ citation needed ] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v w Johnson, RW; Rice, AS (October 2014). ... External links [ edit ] Classification D ICD - 10 : B02.2, G53.0 , G44.847 Mm ICD - 9-CM : 053.19 MeSH : D051474 External resources MedlinePlus : 007423 eMedicine : neuro/317 Patient UK : Postherpetic neuralgia v t e Varicella zoster Varicella zoster virus Varicellovirus Diseases Chickenpox Herpes zoster Postherpetic neuralgia Ramsay Hunt syndrome type II Disseminated herpes zoster Progressive outer retinal necrosis Ophthalmic zoster Treatment Aciclovir Vidarabine VZV immune globulin Prevention Varicella vaccine Zoster vaccine Pox party Other Michiaki Takahashi v t e Artificial induction of immunity / Immunization : Vaccines , Vaccination , Infection , Inoculation ( J07 ) Development Adjuvants List of vaccine ingredients Mathematical modelling Timeline Trials Classes Conjugate vaccine Inactivated vaccine Live vector vaccine Attenuated vaccine Heterologous vaccine Subunit/component / Peptide / Virus-like particle / Synthetic DNA / RNA Toxoid Administration Global: GAVI Alliance NITAG Policy Schedule Vaccine injury US: ACIP Vaccine court Vaccines for Children Program VAERS VSD Vaccines Bacterial Anthrax Brucellosis Cholera # Diphtheria # Hib # Leptospirosis Lyme disease ‡ Meningococcus # MeNZB NmVac4-A/C/Y/W-135 Pertussis # Plague Pneumococcal # PCV PPSV Q fever Tetanus # Tuberculosis BCG # Typhoid # Ty21a ViCPS Typhus combination: DPT/DTwP/DTaP Td/Tdap research: Clostridioides difficile Group B streptococcal disease Shigellosis Viral Adenovirus COVID-19 Oxford–AstraZeneca † Moderna † Pfizer-BioNTech † Ebola Flu # H1N1 ( Pandemrix ) H5N1 LAIV Hantavirus Hepatitis A # Hepatitis B # Hepatitis E HPV Cervarix Gardasil Japanese encephalitis # Measles # Mumps # Mumpsvax Polio # Sabin Salk Rabies # Rotavirus # Rubella # Smallpox Dryvax Tick-borne encephalitis Varicella zoster Chicken pox # Shingles Yellow fever # combination: Hepatitis A and B MMR MMRV research: Chikungunya COVID-19 Cytomegalovirus Dengue Ebola Epstein–Barr virus Hepatitis C Herpes simplex HIV Respiratory syncytial virus Zika Protozoan Malaria RTS,S research: Trypanosomiasis Helminthiasis research: Hookworm Schistosomiasis Other Androvax (androstenedione albumin) Cancer vaccines ALVAC-CEA BCG # Hepatitis B # HPV Cervarix Gardasil Prostvac NicVAX Ovandrotone albumin (Fecundin) TA-CD TA-NIC combination: DTaP-IPV/Hib DTaP-IPV-HepB Hexavalent vaccine Pentavalent vaccine Inventors/researchers Edward Jenner Louis Pasteur Hilary Koprowski Jonas Salk John Franklin Enders Maurice Hilleman Stanley Plotkin H. ... Secretary of Health and Human Services Alternative vaccination schedule Related Epidemiology Eradication of infectious diseases Every Child by Two List of vaccine topics # WHO-EM ‡ Withdrawn from market Clinical trials : † Phase III § Never to phase III v t e Headache Primary ICHD 1 Migraine Familial hemiplegic Retinal migraine ICHD 2 Tension Mixed tension migraine ICHD 3 Cluster Chronic paroxysmal hemicrania SUNCT ICHD 4 Hemicrania continua Thunderclap headache Sexual headache New daily persistent headache Hypnic headache Secondary ICHD 5 Migralepsy ICHD 7 Ictal headache Post-dural-puncture headache ICHD 8 Hangover Medication overuse headache ICHD 13 Trigeminal neuralgia Occipital neuralgia External compression headache Cold-stimulus headache Optic neuritis Postherpetic neuralgia Tolosa–Hunt syndrome Other Vascular
Overview Postherpetic neuralgia (post-hur-PET-ik noo-RAL-juh) is the most common complication of shingles. It causes a burning pain in nerves and skin. The pain lasts long after the rash and blisters of shingles go away. The risk of postherpetic neuralgia rises with age. It mainly affects people older than 60. There's no cure, but treatments can ease symptoms. For most people, postherpetic neuralgia gets better over time. Symptoms In general, the symptoms of postherpetic neuralgia are limited to the area of skin where the shingles outbreak first happened.
In China, the overall five-year survival rate for advanced esophageal cancer is about 20%, and in the United States it is about 15%. [4] Stomach cancer [ edit ] Main article: Gastric cancer Cancer of the stomach , also called gastric cancer, is the fourth-most-common type of cancer and the second-highest cause of cancer death globally. [2] Eastern Asia (China, Japan, Korea, Mongolia) is a high-risk area for gastric cancer, and North America, Australia, New Zealand and western and northern Africa are areas with low risk. [5] The most common type of gastric cancer is adenocarcinoma , which causes about 750,000 deaths each year. [6] Important factors that may contribute to the development of gastric cancer include diet, smoking and alcohol consumption, genetic aspects (including a number of heritable syndromes) and infections (for example, Helicobacter pylori or Epstein-Barr virus ) and pernicious anemia . [1] [6] Chemotherapy improves survival compared to best supportive care, however the optimal regimen is unclear. [7] Pancreatic cancer [ edit ] Upper and lower human gastrointestinal tract Main article: Pancreatic cancer Pancreatic cancer is the fifth-most-common cause of cancer deaths in the United States, [8] and the seventh most common in Europe. [9] In 2008, globally there were 280,000 new cases of pancreatic cancer reported and 265,000 deaths. [10] These cancers are classified as endocrine or nonendocrine tumors. ... If a younger person gets such a cancer, it is often associated with hereditary syndromes like Peutz-Jegher's , hereditary nonpolyposis colorectal cancer or familial adenomatous polyposis . [12] : 619–620 Colorectal cancer can be detected through the bleeding of a polyp , colicky bowel pain, a bowel obstruction or the biopsy of a polyp at a screening colonoscopy . ... External links [ edit ] Gastrointestinal (GI) Cancer Resource Centre Gastrointestinal Cancer Treatment Classification D ICD - 10 : C15 - C26 v t e Digestive system neoplasia GI tract Upper Esophagus Squamous cell carcinoma Adenocarcinoma Stomach Gastric carcinoma Signet ring cell carcinoma Gastric lymphoma MALT lymphoma Linitis plastica Lower Small intestine Duodenal cancer Adenocarcinoma Appendix Carcinoid Pseudomyxoma peritonei Colon/rectum Colorectal polyp : adenoma , hyperplastic , juvenile , sessile serrated adenoma , traditional serrated adenoma , Peutz–Jeghers Cronkhite–Canada Polyposis syndromes: Juvenile MUTYH-associated Familial adenomatous / Gardner's Polymerase proofreading-associated Serrated polyposis Neoplasm: Adenocarcinoma Familial adenomatous polyposis Hereditary nonpolyposis colorectal cancer Anus Squamous cell carcinoma Upper and/or lower Gastrointestinal stromal tumor Krukenberg tumor (metastatic) Accessory Liver malignant : Hepatocellular carcinoma Fibrolamellar Hepatoblastoma benign : Hepatocellular adenoma Cavernous hemangioma hyperplasia : Focal nodular hyperplasia Nodular regenerative hyperplasia Biliary tract bile duct : Cholangiocarcinoma Klatskin tumor gallbladder : Gallbladder cancer Pancreas exocrine pancreas : Adenocarcinoma Pancreatic ductal carcinoma cystic neoplasms : Serous microcystic adenoma Intraductal papillary mucinous neoplasm Mucinous cystic neoplasm Solid pseudopapillary neoplasm Pancreatoblastoma Peritoneum Primary peritoneal carcinoma Peritoneal mesothelioma Desmoplastic small round cell tumor
. ^ Goodlin R C (1985) Pregnant women with Munchausen syndrome. American Journal of Obstetrics and Gynecology 153: 207-210. ^ Jureidini J (1993) Obstetric factitious Disorder and Munchausen syndrome by proxy. ... Psychosomatics 55: 392-395. ^ Feldman F D, Hamilton J C (2006) Serial factitious disorder and Munchausen syndrome by proxy in pregnancy. International Journal of Clinical Practice 60: 1675-1678. ^ name="Cambridge 2017, p94-100." ^ Kirkland T (1774) Treatise on childbed fevers and on the methods of preventing them.
A rare bacterial infectious disease caused by the tick-borne bacterium Rickettsia africae , characterized by acute onset of fever accompanied by myalgia, localized lymphadenitis, and a papulovesicular rash. In most cases at least one, sometimes multiple, inoculation eschars are observed. Clustering of cases is frequent.
Overview Encephalitis (en-sef-uh-LIE-tis) is inflammation of the brain. There are several causes, including viral infection, autoimmune inflammation, bacterial infection, insect bites and others. When inflammation is caused by an infection in the brain, it's known as infectious encephalitis. And when it's caused by your own immune system attacking the brain, it's known as autoimmune encephalitis. Sometimes there is no known cause. In some cases, encephalitis can be life-threatening.
Syndrome of disordered eating, oligomenorrhoea and osteopenia This article needs to be updated . Please update this article to reflect recent events or newly available information. ( December 2018 ) Relative energy deficiency in sport ( RED-S ) [1] [2] is a syndrome in which disordered eating (or low energy availability ), [3] amenorrhoea /oligomenorrhoea, and decreased bone mineral density ( osteoporosis and osteopenia ) are present. [4] It is caused by eating too little food to support the amount of energy being expended by an athlete, often at the urging of a coach or other authority figure who believes that athletes are more likely to win competitions when they have an extremely lean body type. RED-S is a serious illness with lifelong health consequences and can potentially be fatal. [5] RED-S is the broader, more comprehensive name for what was formerly known as female athlete triad (or simply the triad ), which was a condition seen in females participating in sports that emphasize leanness or low body weight. [1] [6] As the non-menstrual components are also seen in males, the name was changed to the comprehensive term RED-S. [1] Contents 1 Classification 2 Signs and symptoms 2.1 Disordered eating 2.2 Amenorrhea 2.3 Osteoporosis 3 Causes 4 Treatment 4.1 Less exercise 4.2 Eating more 4.3 Medicine 5 Prognosis 6 Society and culture 7 See also 8 References 9 External links Classification [ edit ] Formerly known as the female athlete triad, RED-S is a syndrome of three interrelated conditions.
An earworm , sometimes referred to as a brainworm , [1] sticky music , stuck song syndrome , [2] or, most commonly after earworms, Involuntary Musical Imagery (INMI) , [3] [4] [5] [6] [7] is a catchy and/or memorable piece of music or saying that continuously occupies a person's mind even after it is no longer being played or spoken about. [8] [9] Involuntary musical imagery as a label is not solely restricted to earworms; musical hallucinations also fall into this category, although they are not the same thing. [4] [10] Earworms are considered to be a common type of involuntary cognition . [11] Some of the phrases often used to describe earworms include "musical imagery repetition" and "involuntary musical imagery". [1] [12] [13] The word earworm is a calque from the German Ohrwurm , which has had this sense since the mid-20th century. [14] [15] The earliest [ citation needed ] known English usage is in Desmond Bagley 's 1978 novel Flyaway , where the author points out the German origin of his coinage. [16] Researchers who have studied and written about the phenomenon include Theodor Reik , [17] Sean Bennett, [18] Oliver Sacks , [1] Daniel Levitin , [19] James Kellaris, [20] Philip Beaman, [21] Vicky Williamson, [22] Diana Deutsch , [23] and, in a more theoretical perspective, Peter Szendy , [24] along with many more. ... Scottsdale, Arizona: American Psychological Society : 66–67. ^ a b Beaman CP, Williams TI (November 2010). "Earworms (stuck song syndrome): towards a natural history of intrusive thoughts" . ... Philip; Williams, Tim I. (2010). "Earworms (stuck song syndrome): Towards a natural history of intrusive thoughts" .
Tropical Race 4 (TR4) was first identified in Taiwan, [2] and from there rapidly spread to Indonesia, China, Malaysia, Australia and the Philippines. [3] The disease was then identified in Jordan in 2013. [4] TR4 later spread to Vietnam [5] and Laos, [6] as well as to the Middle East being reported in Pakistan and Lebanon. [7] In 2015, the disease then spread to Africa, being informally announced in Mozambique and Oman. [3] In August 2019, TR4 arrived in Colombia , a country in Latin America , the region comprising the world's biggest banana exporters. [8] Symptoms [ edit ] Two external symptoms help characterize Panama disease of banana: Yellow leaf syndrome, the yellowing of the border of the leaves which eventually leads to bending of the petiole. [1] Green leaf syndrome, which occurs in certain cultivars, marked by the persistence of the green color of the leaves followed by the bending of the petiole as in yellow leaf syndrome.
Eastern equine encephalitis (Triple E) is an illness caused by a virus that comes from an infected mosquito bite. Most people who are infected have no symptoms, but some may develop fever, headaches, and vomiting. Symptoms occur within 4-10 days of being infected and most recover in 1-2 weeks. A small number will develop swelling of the brain and severe neurological symptoms that can be permanent. Triple E can also lead to coma and death. This condition is caused by a type of arbovirus and is transmitted by a mosquito which has bitten an infected bird or reptile.
An acute arboviral infection caused by an alphavirus of the Togaviridae family transmitted by an infected mosquito, that is characterized by the onset of flulike symptoms including fever, chills, weakness, headache, vomiting, abdominal pain with diarrhea, myalgia, leucocytosis, and hematuria, rapidly progressing to diffuse central nervous system (CNS) involvement with confusion, somnolence, or even coma. Seizures, which may progress to status epilepticus and neurologic sequelae, cranial nerve palsies, and photophobia may occur. EEE is associated with a high rate of morbidity and mortality.
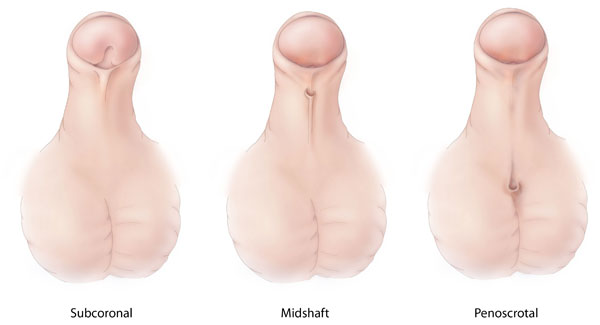
It most often occurs by itself, without other variations, although in about 10% of cases it may be part of an intersex condition or a medical syndrome with multiple abnormalities. [6] [7] The most common associated difference is an undescended testicle , which has been reported in around 3% of infants with distal hypospadias and 10% with proximal hypospadias. [8] The combination of hypospadias and an undescended testicle sometimes indicates a child has an intersex condition, so additional testing may be recommended to make sure the child does not have congenital adrenal hyperplasia with salt wasting or a similar condition where immediate medical intervention is needed. [9] [10] Otherwise no blood tests or X-rays are routinely needed in newborns with hypospadias. [3] Hypospadias can be a symptom or indication of an intersex condition, but the presence of hypospadias alone is not enough to classify a person as intersex. ... Martín [34] [35] Scout Schultz [36] See also [ edit ] Pediatric urology Andrology Cryptorchidism Bladder exstrophy , cloacal exstrophy Perineal urethra , pseudovaginal perineoscrotal hypospadias Ambiguous genitalia , intersex , intersex surgery Androgen insensitivity syndrome Testicular dysgenesis syndrome References [ edit ] ^ Entry "hypospadias" in Merriam-Webster Online Dictionary . ^ OED 2nd edition, 1989 as /hɪpəʊˈspeɪdɪəs/ ~ /haɪpəʊˈspeɪdɪəs/ ^ a b c d e Snodgrass W (2012).
Overview Hypospadias (hi-poe-SPAY-dee-us) is a birth defect (congenital condition) in which the opening of the urethra is on the underside of the penis instead of at the tip. The urethra is the tube through which urine drains from your bladder and exits your body. Hypospadias is common and doesn't cause difficulty in caring for your infant. Surgery usually restores the normal appearance of your child's penis. With successful treatment of hypospadias, most males can have normal urination and reproduction.
In secondary hyperparathyroidism due to lack of Vitamin D absorption, the parathyroid gland is behaving normally; clinical problems are due to bone resorption and manifest as bone syndromes such as rickets , osteomalacia , and renal osteodystrophy . [12] Causes [ edit ] Radiation exposure increases the risk of primary hyperparathyroidism. [1] A number of genetic conditions including multiple endocrine neoplasia syndromes also increase the risk. [1] Mechanism [ edit ] Normal parathyroid glands measure the ionized calcium (Ca 2+ ) concentration in the blood and secrete parathyroid hormone accordingly; if the ionized calcium rises above normal, the secretion of PTH is decreased, whereas when the Ca 2+ level falls, parathyroid hormone secretion is increased. [8] Secondary hyperparathyroidism occurs if the calcium level is abnormally low. ... This disease is often characterized by the quartet stones, bones, groans, and psychiatric overtones referring to the presence of kidney stones , hypercalcemia, constipation, and peptic ulcers , as well as depression , respectively. [17] [18] In a minority of cases, this occurs as part of a multiple endocrine neoplasia (MEN) syndrome, either type 1 (caused by a mutation in the gene MEN1 ) or type 2a (caused by a mutation in the gene RET ), which is also associated with the adrenal tumor pheochromcytoma .
Overview Hyperparathyroidism is when your parathyroid glands create high amounts of parathyroid hormone in the bloodstream. These glands, located behind the thyroid at the bottom of your neck, are about the size of a grain of rice. Parathyroid glands The parathyroid glands lie behind the thyroid. They produce parathyroid hormone, which plays a role in regulating the body's blood level of calcium and phosphorus. The parathyroid hormone produced by the thyroid glands helps maintain the right balance of calcium in the bloodstream and in tissues that depend on calcium for proper functioning. This is especially important for nerve and muscle function, as well as bone health.
These conditions include ulcerative colitis and Crohn's disease. Inherited syndromes that increase colon cancer risk. Some DNA changes that increase the risk of colon cancer run in families. The most common inherited syndromes that increase colon cancer risk are familial adenomatous polyposis and Lynch syndrome.