However, the association with chromosome 22q11 deletion ( DiGeorge Syndrome ) implies that a genetic component is likely in certain cases. [3] Esophageal atresia also occurs in some patients with double aortic arch. [4] Diagnosis [ edit ] Prenatal diagnosis (fetal ultrasound): Today the diagnosis of double aortic arch can be obtained in-utero in experienced centers. [5] Scheduled repair soon after birth in symptomatic patients can relieve tracheal compression early and therefore potentially prevent the development of severe tracheomalacia. ... J Am Coll Cardiol. 1995;25:943 External links [ edit ] Classification D DiseasesDB : 33811 External resources MedlinePlus : 007316 eMedicine : article/899609 v t e Congenital vascular defects / Vascular malformation Great arteries / other arteries Aorta Patent ductus arteriosus Coarctation of the aorta Interrupted aortic arch Double aortic arch Right-sided aortic arch Overriding aorta Aneurysm of sinus of Valsalva Vascular ring Pulmonary artery Pulmonary atresia Stenosis of pulmonary artery Subclavian artery Aberrant subclavian artery Umbilical artery Single umbilical artery Great veins Superior / inferior vena cava Congenital stenosis of vena cava Persistent left superior vena cava Pulmonary vein Anomalous pulmonary venous connection ( Total , Partial ) Scimitar syndrome Arteriovenous malformation Cerebral arteriovenous malformation
People with this type of aphasia often have trouble understanding other's speech and generally do not realize that they are not making any sense. [16] Conduction aphasia [16] Anomic aphasia [16] Global aphasia [16] Primary progressive aphasias Progressive nonfluent aphasia [17] Semantic dementia [17] Logopenic progressive aphasia [17] Learning disability [ edit ] Dyscalculia – a defect of the systems used in communicating numbers Dyslexia – a defect of the systems used in reading Dysgraphia – a defect in the systems used in writing Speech disorders [ edit ] cluttering - a syndrome characterized by a speech delivery rate which is either abnormally fast, irregular, or both. [18] dysarthria - a condition that occurs when problems with the muscles that helps a person to talk make it difficult to pronounce words. [19] esophageal voice - involves the patient injecting or swallowing air into the esophagus. ... External links [ edit ] Classification D ICD - 10 : F80 ICD - 9-CM : 315.3 MeSH : D003147 SNOMED CT : 278919001 Communication Disorders Aphasia - National Institute on Deafness and Other Communication Disorders (NIDCD) Dysgraphia - National Institute on Deafness and Other Communication Disorders Voice and Speech Disorder Online Community (VoiceMatters.net) List of communication disorder related links Child Language Disorders Talking Point Check the progress of your child's language development Topics related to Communication disorder v t e Dyslexia and related specific developmental disorders Conditions Speech, language , and communication Expressive language disorder Infantile speech Landau–Kleffner syndrome Language disorder Lisp Mixed receptive-expressive language disorder Specific language impairment Speech and language impairment Speech disorder Speech error Speech sound disorder Stuttering Tip of the tongue Learning disability Dyslexia Dyscalculia Dysgraphia Disorder of written expression Motor Developmental coordination disorder Developmental verbal dyspraxia Sensory Auditory processing disorder Sensory processing disorder Related topics Dyslexia research Irlen filters Learning Ally Learning problems in childhood cancer Literacy Management of dyslexia Multisensory integration Neuropsychology Reading acquisition Spelling Writing system Lists Dyslexia in fiction Languages by Writing System People with dyslexia
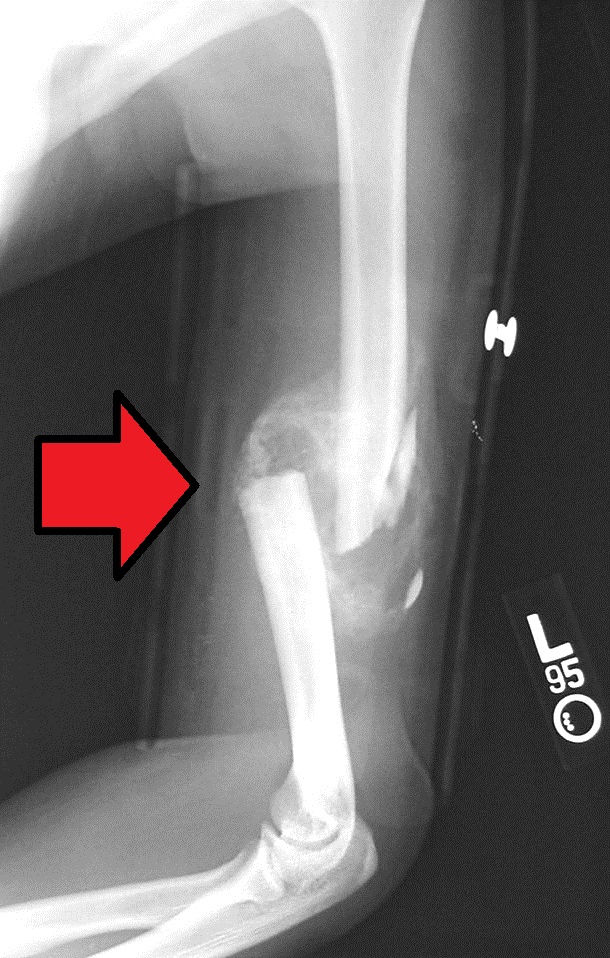
Humerus fracture Midshaft humerus fracture with callus formation Specialty Orthopedics Symptoms Pain, swelling, bruising [1] Complications Injury to an artery or nerve, compartment syndrome [2] Types Proximal humerus , humerus shaft, distal humerus [1] [2] Causes Trauma , cancer [2] Diagnostic method X-rays [2] Treatment Sling , splint, brace , surgery [1] Prognosis Generally good (proximal and shaft), Less good (distal) [2] Frequency ~4% of fractures [2] A humerus fracture is a break of the humerus bone in the upper arm . [1] Symptoms may include pain, swelling, and bruising . [1] There may be a decreased ability to move the arm and the person may present holding their elbow. [2] Complications may include injury to an artery or nerve, and compartment syndrome . [2] The cause of a humerus fracture is usually physical trauma such as a fall . [1] Other causes include conditions such as cancer in the bone. [2] Types include proximal humeral fractures , humeral shaft fractures, and distal humeral fractures . [1] [2] Diagnosis is generally confirmed by X-rays . [2] A CT scan may be done in proximal fractures to gather further details. [2] Treatment options may include a sling , splint, brace , or surgery. [1] In proximal fractures that remain well aligned, a sling is often sufficient. [2] Many humerus shaft fractures may be treated with a brace rather than surgery. [2] Surgical options may include open reduction and internal fixation , closed reduction and percutaneous pinning , and intramedullary nailing . [2] Joint replacement may be another option. [2] Proximal and shaft fractures generally have a good outcome while outcomes with distal fractures can be less good. [2] They represent about 4% of fractures. [2] Contents 1 Signs and symptoms 2 Causes 2.1 Proximal 2.2 Middle 2.3 Distal 3 Diagnosis 3.1 Classification 4 Treatment 4.1 Proximal 4.2 Middle 5 Prognosis 6 Epidemiology 7 References 7.1 Bibliography 8 External links Signs and symptoms [ edit ] Closed shaft fracture in the middle of humerus After a humerus fracture, pain is immediate, enduring, and exacerbated with the slightest movements.
They suggested that this approach may be particularly useful for rare X-linked disorders such as Lowe syndrome and Hunter syndrome, that the recombinant X chromosomes should be maintained as fibroblasts or lymphoblastoid cells in cell repositories, and that the approach is also useful in autosomal mapping.
A rare, genetic muscular dystrophy characterized by progressive muscle wasting and weakness due to degeneration of skeletal, smooth and cardiac muscle. Epidemiology Becker muscular dystrophy (BMD) primarily affects males; in Europe the estimated prevalence ranges between 1:16,700-1:18,500 male births. Clinical description Onset is usually in childhood, typically after 7 years of age, but can be later. Presenting features in children include toe walking gait and or exercise-related cramps with or without myoglobinuria. Some patients may present following anesthetic induced acute rhabdomyolysis.
Hastings et al. (1980) described 2 unrelated families, each with father and son with pseudohypertrophic muscular dystrophy. The paternal grandfather in 1 family may have been affected also. The phenotype resembled that of Becker muscular dystrophy (300376). The mothers showed no evidence of carrier status, but both fathers had pseudohypertrophic calves and one gave a history of weakness in childhood with subsequent improvement. Muscle histology in all 4 showed changes like those of Becker muscular dystrophy with, in addition, central cores and internalized capillaries in type I fibers. The internalized capillaries were considered unique to this disorder.
Muscular dystrophies are a group of genetic conditions characterized by progressive muscle weakness and wasting (atrophy). The Duchenne and Becker types of muscular dystrophy are two related conditions that primarily affect skeletal muscles , which are used for movement, and heart (cardiac) muscle. These forms of muscular dystrophy occur almost exclusively in males. Duchenne and Becker muscular dystrophies have similar signs and symptoms and are caused by different mutations in the same gene. The two conditions differ in their severity, age of onset, and rate of progression. In boys with Duchenne muscular dystrophy, muscle weakness tends to appear in early childhood and worsen rapidly.
A group of rare, genetic, progressive muscular dystrophies, including Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD) and a symptomatic form in female carriers. The diseases represent a spectrum of severity ranging from progressive skeletal and cardiac muscle wasting and weakness (DMD, BMD) to less severe muscle weakness or isolated cardiomyopathy affecting carrier females. Epidemiology The prevalence of DMD ranges between 1/3,500-1/ 9,300 male births. The prevalence of BMD varies from 1/16,700 to 1/18,500 male births. The prevalence of symptomatic female carriers is unknown. Clinical description Dystrophinopathies present with a spectrum of severity whereby BMD is at the mildest end and DMD the most severe, there is an intermediate phenotype in between.
Becker muscular dystrophy (BMD) is an inherited condition that causes progressive weakness and wasting of the skeletal and cardiac (heart) muscles. It primarily affects males. The age of onset and rate of progression can vary. Muscle weakness usually becomes apparent between the ages of 5 and 15. In some cases, heart involvement (cardiomyopathy) is the first sign. BMD is caused by a mutation in the DMD gene and is inherited in an X-linked recessive manner. BMD is very similar to Duchenne muscular dystrophy , except that in BMD, symptoms begin later and progress at a slower rate.
Burwinkel et al. (2005) noted that this severe phenotype characterized by fetal onset, extreme cardiomegaly, and death in infancy extended the clinical spectrum of PRKAG2 mutations, which had previously been shown to cause familial hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome (600858). Patients with the R531Q mutation died of hemodynamic and respiratory failure secondary to hypertrophic nonobstructive cardiomyopathy, but also had Wolff-Parkinson-White-like conduction anomalies.
Additional features that occur in people with APECED, many of which can lead to permanent organ and tissue damage if left untreated, include stomach irritation (gastritis), liver inflammation (hepatitis), lung irritation (pneumonitis), dry mouth and dry eyes (Sjogren-like syndrome), inflammation of the eyes (keratitis), kidney problems (nephritis), vitamin B12 deficiency, hair loss (alopecia), loss of skin color in blotches (vitiligo), high blood pressure (hypertension), or a small (atrophic) or absent spleen (asplenia).
Because he had abnormal lower incisors and a bifid uvula, Nance-Horan syndrome (NHS; 302350) was suspected, but analysis of the NHS gene (300457) was negative.
A rare, genetic form of pontocerebellar hypoplasia (PCH) characterized by neocortical and severe cerebral cortical atrophy associated with pontocerebellar hypoplasia with the pons and cerebellum equally affected. Clinically the disorder manifests at birth with hypotonia, clonus, epilepsy impaired swallowing and from infancy by progressive microcephaly, spasticity and lactic acidosis. Epidemiology Pontocerebellar hypoplasia type 6 (PCH6) is reported in at least 31 cases to date. Clinical description PCH6 manifests at birth by generalized hypotonia, lethargy and dysphagia. The clinical profile is characterized from infancy by a profound developmental delay, progressive microcephaly, hypotonia or spasticity and treatment-resistant epilepsy.
A number sign (#) is used with this entry because X-linked VACTERL syndrome with or without hydrocephalus (VACTERLX) is caused by mutation in or deletion of the ZIC3 gene (300265) on chromosome Xq26.
Description VACTERL describes a constellation of congenital anomalies, including vertebral anomalies, anal atresia, congenital cardiac disease, tracheoesophageal fistula, renal anomalies, radial dysplasia, and other limb defects; see 192350. Cases of familial VACTERL with hydrocephalus (H) have been reported with suggestion of autosomal recessive or X-linked inheritance (see 314390). Other patients thought to have VACTERL-H, including 2 unrelated infants reported by Porteous et al. (1992), had been found to have Fanconi anemia (see 227650). Porteous et al. (1992) suggested that chromosomal breakage studies should be performed in all cases of VACTERL/VACTERL-H to rule out Fanconi anemia. Alter et al. (2007) noted that a VATER phenotype had been reported in Fanconi anemia of complementation groups A (227650), C (227645), D1 (605724), E (600901), F (603467), and G (614082).
VACTERL is an acronym for Vertebral anomalies, Anal atresia, Congenital cardiac disease, Tracheoesophageal fistula, Renal anomalies, and Limb defects. VACTERL associated with hydrocephalus has rarely been reported and is thought to be an autosomal recessive anomaly. The condition is described as a uniformly lethal or developmentally devastating disorder distinct from the VATER association.
Familial aggregation in this condition is probably of the same order and same basis as that of many autoimmune disorders (109100) such as Hashimoto struma (140300), Schmidt syndrome (269200), autoimmune hemolytic anemia (205700), lupus erythematosus (152700), alopecia areata (104000), pernicious anemia (170900), etc.
Overview Giant cell arteritis is an inflammation of the lining of your arteries. Most often, it affects the arteries in your head, especially those in your temples. For this reason, giant cell arteritis is sometimes called temporal arteritis. Giant cell arteritis frequently causes headaches, scalp tenderness, jaw pain and vision problems. Untreated, it can lead to blindness. Prompt treatment with corticosteroid medications usually relieves symptoms of giant cell arteritis and might prevent loss of vision.
Giant cell arteritis (GCA) is a large vessel vasculitis predominantly involving the arteries originating from the aortic arch and especially the extracranial branches of the carotid arteries. Epidemiology GCA is the most common adulthood vasculitis with an annual incidence of 1/3,000-1/25,000 adults over 50 years old. It is more frequent in populations of northern European background. GCA affects people of more than 50 years old (median age at diagnosis between 70-75 years old) and occurs twice as frequently in women as in men. Clinical description GCA often starts insidiously with general symptoms, cranial manifestations (headache, jaw claudication, scalp tenderness, visual loss), and, in about 50% of patients, polymyalgia rheumatica. Visual symptoms due to an ischemic optic neuropathy occur in 20-30% of patients, and can rapidly lead to irreversible monocular blindness.
Giant cell arteritis (GCA) is a form of vasculitis , a group of disorders that cause inflammation of blood vessels. GCA most commonly affects the arteries of the head (especially the temporal arteries, located on each side of the head), but arteries in other areas of the body can also become inflamed. The inflammation causes the arteries to narrow, resulting in poor blood flow. Signs and symptoms when arteries in the head are involved may include a throbbing headache on one side or the back of the head, tenderness of the scalp, flu-like symptoms, and/or problems with eyesight. Symptoms when other arteries are involved depend on the location of those arteries.
Over the next 2 years, he had recurrent palpitations and chest pain with normal angiography, and was diagnosed with Wolff-Parkinson-White preexcitation syndrome (WPW; see 194200). He eventually underwent placement of an internal cardioverter-defibrillator for symptomatic nonsustained ventricular tachycardia.
Garg et al. (2015) reported 2 unrelated children, a 7-year-old boy and a 3-year-old girl, with a complex lipodystrophy syndrome apparent from birth. As infants, they presented with loss of subcutaneous fat, but not from the buttocks, and thin skin with prominent vessels suggestive of cutis marmorata.
The disorder shows phenotypic similarities to Kohlschutter-Tonz syndrome (KTZS; 226750), which is caused by mutation in the mutation in the ROGDI gene (614574) on chromosome 16p13.