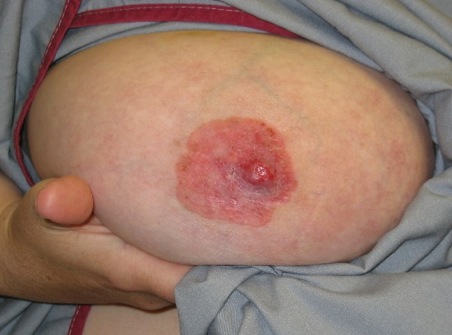
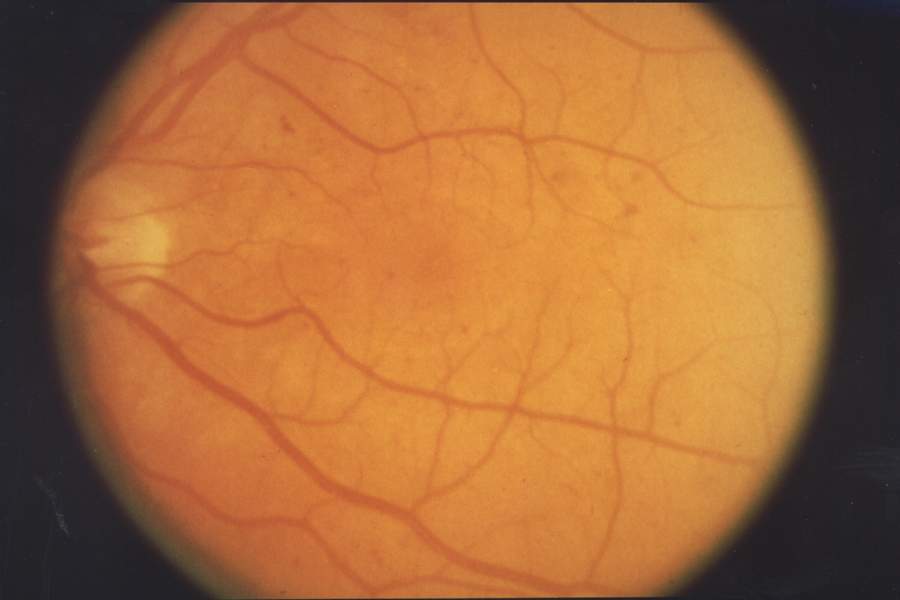
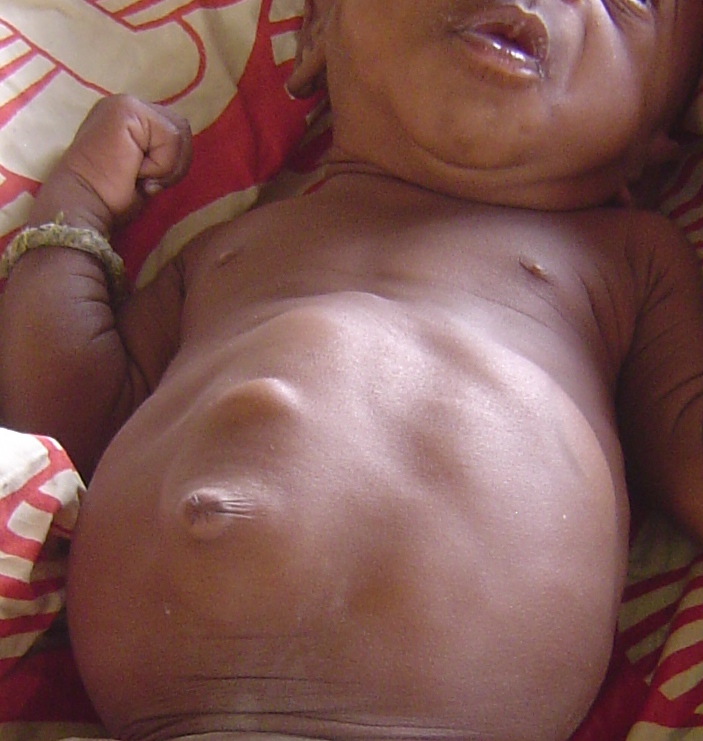
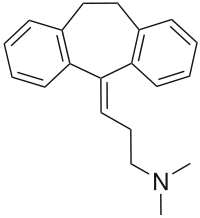
Two SNPs were significantly associated with major depressive syndrome (p = 0.004 and 0.017). Haplotype analyses revealed that the most common haplotype, T-T-T (rs1824024, rs2061174, and rs324650), was undertransmitted to affected individuals with alcohol dependence and major depressive syndrome.
Depression (also known as major depression or major depressive disorder) is a psychiatric disorder that affects mood, behavior, and overall health. It causes prolonged feelings of sadness, emptiness, or hopelessness, and a loss of interest in activities that were once enjoyed. People with depression may also have changes in appetite (leading to overeating or not eating enough), changes in sleeping patterns (sleeping too much or not being able to sleep), loss of energy, and difficulty concentrating. Although depression is considered primarily a mental health disorder, it can also have physical features including headaches, other unexplained aches and pains, unusually slow or fast movements, and digestive problems. To be diagnosed with depression, an individual must have signs and symptoms nearly every day for at least 2 weeks.
Seasonal affective disorder is a mental health condition that is triggered by the changing of the seasons. This condition is a subtype of major depressive disorder and bipolar disorder. Major depressive disorder is characterized by prolonged sadness and a general lack of interest, while bipolar disorder is characterized by similar depressive episodes alternating with periods of abnormally high energy and activity (hypomania or mania). People with seasonal affective disorder have signs and symptoms of either major depressive disorder or bipolar disorder only during certain months of the year. Major depressive disorder is more common than bipolar disorder among people with seasonal affective disorder.
Wernicke encephalopathy and Korsakoff syndrome are forms of beriberi. Alcoholism can also cause vitamin deficiency. ... Symptoms include irritability, fatigue, and apathy . [24] [25] Vitamin B 6 deficiency is uncommon, although it may be observed in certain conditions, such as end-stage kidney diseases or malabsorption syndromes , such as celiac disease , Crohn disease or ulcerative colitis . ... External links [ edit ] Classification D ICD - 10 : E50-E56 ICD - 9-CM : 264 - 269 MeSH : D001361 v t e Malnutrition Protein-energy malnutrition Kwashiorkor Marasmus Catabolysis Vitamin deficiency B vitamins B 1 Beriberi Wernicke–Korsakoff syndrome Wernicke's encephalopathy Korsakoff's syndrome B 2 Riboflavin deficiency B 3 Pellagra B 6 Pyridoxine deficiency B 7 Biotin deficiency B 9 Folate deficiency B 12 Vitamin B 12 deficiency Other A: Vitamin A deficiency Bitot's spots C: Scurvy D: Vitamin D deficiency Rickets Osteomalacia Harrison's groove E: Vitamin E deficiency K: Vitamin K deficiency Mineral deficiency Sodium Potassium Magnesium Calcium Iron Zinc Manganese Copper Iodine Chromium Molybdenum Selenium Keshan disease Growth Delayed milestone Failure to thrive Short stature Idiopathic General Anorexia Weight loss Cachexia Underweight
., SMN1 copy number analysis, methylation testing for Prader-Willi syndrome, UPD14 analysis) is recommended (see Table 1). (2) Although the CTG repeat expansion will not be detected by a multigene sequencing panel , this testing may be appropriate for some conditions in the differential diagnosis. (3) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (4) Some multigene panels may include genes not associated with the condition discussed in this GeneReview . (5) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. ... Differential Diagnosis Hypotonia in infancy is seen in many disorders, including Prader-Willi syndrome, multiminicore disease (OMIM 606210, 180901), nemaline myopathy (OMIM PS161800), X-linked centronuclear myopathy, other centronuclear myopathies (OMIM PS160150), and maternal uniparental disomy for chromosome 14.
This may be a situation like that of the fragile X syndrome in which rare affected individuals lack a trinucleotide repeat expansion and instead have deletions or point mutations. ... They suggested that the mutational mechanism leading to DM is triplet repeat amplification, similar to that occurring in the fragile X syndrome. The genomic repeat is p(AGC)n.
Myotonic dystrophy type 1 (MD1), one of the two types of myotonic dystrophy , is an inherited type of muscular dystrophy that affects the muscles and other body systems (e.g., heart, eyes, endocrine system , and central nervous system). MD1 has three forms that somewhat overlap: the mild form, classic form, and congenital form (present at birth). The mild form has the least severe symptoms of the different forms of MD1 and is associated with a normal life span. The classic form is characterized by muscle weakness and wasting, prolonged muscle tensing ( myotonia ), cataract, and often, abnormal heart function. Adults with the classic form may become physically disabled and may have a shortened life span.
Myotonic dystrophy is part of a group of inherited disorders called muscular dystrophies. It is the most common form of muscular dystrophy that begins in adulthood. Myotonic dystrophy is characterized by progressive muscle wasting and weakness. People with this disorder often have prolonged muscle contractions (myotonia) and are not able to relax certain muscles after use. For example, a person may have difficulty releasing their grip on a doorknob or handle.
A rare genetic multi-system disorder characterized by a wide range of muscle-related manifestations (muscle weakness, myotonia, early onset cataracts (before age 50) and systemic manifestations (cerebral, endocrine, cardiac, gastrointestinal tract, uterus, skin and immunologic involvement) that vary depending on the age of onset. The very wide clinical spectrum ranges from lethal presentations in infancy to mild, late-onset disease. Epidemiology It is the most frequent adult muscular dystrophy and has an estimated prevalence ranging from 1/215,000 in Taiwan to 1/5,500 in Croatia. It appears to be more prevalent in the Saguenay-Lac-St-Jean region-Quebec, Canada (1/600), suggesting a founder effect. The disease occurs worldwide. Clinical description The age of onset is highly variable, from prenatal to adulthood.
Differential Diagnosis Congenital (or prelingual) inherited hearing impairment affects approximately one in 1,000 newborns; 30% of these infants have additional anomalies, making the diagnosis of a syndromic form of hearing impairment possible (see Hereditary Deafness and Hearing Loss Overview).
A number sign (#) is used with this entry because autosomal recessive deafness-9 (DFNB9) and auditory neuropathy-1 (AUNB1) are caused by homozygous or compound heterozygous mutation in the gene encoding otoferlin (OTOF; 603681) on chromosome 2p23. Clinical Features Chaib et al. (1996) reported a consanguineous Lebanese family with autosomal recessive sensorineural nonsyndromic hearing loss. For affected children, deafness was noted by their parents at birth or before the age of 2 years. None of the children had balance problems, and there was no evidence for an acquired risk factor predisposing to hearing loss. Audiometry showed no response at 100 dB for frequencies superior to 1,000 Hz in all affected subjects.
The duplication does not lead to a clinically recognizable syndrome, and a subset of individuals with the duplication have no obvious clinical phenotype.
Multiple system atrophy with orthostatic hypotension. Also called Shy-Drager syndrome, this rare disorder affects the nervous system that controls involuntary functions such as blood pressure, heart rate, breathing and digestion.
Description Blue cone (OPN1SW; 613522) monochromatism is a rare X-linked congenital stationary cone dysfunction syndrome characterized by the absence of functional long wavelength-sensitive and medium wavelength-sensitive cones in the retina.
Blue cone monochromatism is an inherited vision disorder. In this condition, the light sensitive cells in the eye used for color vision (cones) are affected. There are three types of cones that respond to one of three colors: red, green, and blue. When people have blue cone monochromatism, both the red and green cones do not function properly, while the blue cones work normally. Signs and symptoms may include impaired color vision, low visual acuity (clarity or sharpness), photophobia (light sensitivity), myopia (nearsightedness), and nystagmus (fast, uncontrollable movements of the eye). Blue cone monochromatism is caused by mutations in either the OPN1LW or the OPN1MW gene(s) and is inherited in an X-linked manner.
Color vision deficiency (sometimes called color blindness) represents a group of conditions that affect the perception of color. Red-green color vision defects are the most common form of color vision deficiency. Affected individuals have trouble distinguishing between some shades of red, yellow, and green. Blue-yellow color vision defects (also called tritan defects), which are rarer, cause problems with differentiating shades of blue and green and cause difficulty distinguishing dark blue from black. These two forms of color vision deficiency disrupt color perception but do not affect the sharpness of vision (visual acuity).
Blue cone monochromatism (BCM) is a recessive X-linked disease characterized by severely impaired color discrimination, low visual acuity, nystagmus, and photophobia, due to dysfunction of the red (L) and green (M) cone photoreceptors. BCM is as an incomplete form of achromatopsia (see this term). Epidemiology The prevalence is estimated to be 1/100,000 worldwide. Clinical description BCM manifests in early infancy and predominantly affects males, with severely impaired color vision and low visual acuity (only rod and blue cone function is preserved). Additionally, photophobia, myopia, and pendular nystagmus are commonly observed. Nystagmus may wane with time. Etiology The disorder is caused by mutations in the red and green opsin gene cluster OPN1LW and OPN1MW (Xq28) and thus affect the corresponding cones.
The marked difference in phenotypic expression is unexplained but is comparable to the difference between Hurler (607014) and Scheie (607016) syndromes, the late infantile and adult forms of metachromatic leukodystrophy (see also 607015), and the classic and visceral forms, A and B (607616), respectively, of Niemann-Pick disease.
Cholesteryl ester storage disease is is a type of lysosomal acid lipase deficiency . It is an inherited disease that causes a buildup of fats (lipids) in the tissues and organs of the body and calcium deposits in the adrenal glands . The liver is most severely affected in most cases. Some people with cholesteryl ester storage disease may develop liver cirrhosis that progresses to liver failure. People with cholesteryl ester storage disease may also build up fatty deposits on the artery walls ( atherosclerosis ). This buildup can narrow the arteries and increase the risk for heart attack or stroke.
A form of lysosomal acid lipase deficiency characterized by progressive cholesterol esters and triglyceride accumulation in tissues and organs typically presenting with hepatosplenomegaly, liver dysfunction and/or dyslipidemia.
But when diarrhea lasts beyond a few days into weeks, it usually indicates that there's another problem — such as irritable bowel syndrome (IBS) or a more serious disorder, including persistent infection, celiac disease or inflammatory bowel disease (IBD).
None of the patients had glaucoma, night blindness, clinical signs of retinal degeneration, developmental ocular malformations, or syndromic disease. Nowilaty et al. (2013) also observed corneal steepening proportional to the degree of axial foreshortening.
Overview Retinal diseases vary widely, but most of them cause visual symptoms. Retinal diseases can affect any part of your retina, a thin layer of tissue on the inside back wall of your eye. The retina contains millions of light-sensitive cells (rods and cones) and other nerve cells that receive and organize visual information. Your retina sends this information to your brain through your optic nerve, enabling you to see. Treatment is available for some retinal diseases. Depending on your condition, treatment goals may be to stop or slow the disease and preserve, improve or restore your vision.
However, Pinel's concept focused on a frenzy of the passions, particularly involving rage and violence. For Prichard the typical syndrome was more a form of extreme eccentricity, and he would later refer to Pinel's type as a madness of the instincts. [7] Prichard was an adherent of what was known faculty psychology , which attempted to divide the mind into different functions or abilities, but not phrenology , which attempted to locate them below specific parts of the skull.
Another possible area of damage leading to IA is the submarginal gyrus , which is located in the parietal lobe of the brain. [8] Overall, IA is an autonomous syndrome, linked to damage in the left hemisphere involving semantic memory disorders rather than a defect in motor control. [9] Several severe injuries or diseases can cause IA in a wide range of patients.
The symptoms can be confused with: hypocalcemia [1] Sepsis [1] CNS disorders [1] Cardiorespiratory problems [1] Neonatal hypoglycemia can also show no symptoms in some newborns or may be life-threatening. [2] Some observed symptoms are (these symptoms may be transient but reoccurring): Jitteriness [2] hypothermia [5] irritability [5] pallor [5] tremors [1] twitching [1] weak or high pitched cry lethargy [2] hypotonia [1] generalized seizures [2] [6] coma [2] cyanosis [2] apnea [2] rapid and irregular respirations [1] diaphoresis [2] eye rolling [1] refusal to feed [1] hunger [2] Complications [ edit ] Long term complications of neonatal hypoglycemia include: Neurologic damage that results in mental retardation [2] Developmental delay [2] Personality disorders [2] Recurrent seizure activity [2] Impaired cardiovascular function [2] Effects of neonatal hypoglycemia [ edit ] Infants that experienced hypoglycemic episodes requiring treatment within the first few days of life have a higher chance of developing neurological or neurodevelopmental diagnoses than normoglycemic infants. [7] The severity of the effects resulting from the hypoglycemic episode depend on the length of the hypoglycemic episode and how low the neonate's blood glucose levels drop during the episode. [8] Because glucose is an essential nutrient for the brain, untreated neonatal hypoglycemia causes irreversible damage to both the posterior occipital and cortex regions of the brain. [9] These areas function in cognition, adaptability, and visual skills. [9] Cause [ edit ] Risks [ edit ] Mother [ edit ] Risk factors in the mother that increased the risk of developing hypoglycemia shortly after birth include: Type 1 diabetes Gestational diabetes mellitus (Transient) [1] Intrapartum glucose administration (Transient) [1] Gestational hypertension [1] Preeclampsia [1] Terbutaline administration (Transient) [1] Intrauterine growth restriction (Transient) [1] Perinatal stress or asphyxia (Transient) [1] Baby [ edit ] Babies which have an increased risk of developing hypoglycemia shortly after birth are: hypothermia (Transient) [1] polycythemia (Transient) [1] hyperinsulinism (Recurrent) [1] IEMs (Recurrent) [1] Beckwith-Wiedmann syndrome (Recurrent) [1] nesidioblastosis (Recurrent) [1] Rh isoimmunization (Recurrent) [1] Certain endocrine disorders (Recurrent) [1] fetal hydrops [1] Prematurity (Transient) [1] congenital malformations [1] small for gestational age (Transient) [1] Large for gestational age (Transient) [1] Hyperinsulinism [ edit ] The most common cause on neonatal hypoglycemia is hyperinsulinism. [2] Hyperinsulinism is also called persistent hyperinsulemic hypoglycemia of infancy (PHHI). [2] This is seen very frequently to the neonates born from mothers with diabetes. [2] Congenital hyperinsulinism is correlated with the abnormality of beta-cell regulation within the pancreas. [2] Isolated islet adenoma, which is a focal disease, is often the cause of congenital hyperinsulism. [2] Drug-induced hyperinsulisim is correlated with the administration insulin or use of hypoglycemic medication. [2] In critical cases, a drug called Diazoxide is availed to stop any secretion of insulin. [2] Limited glycogen stores [ edit ] Limited glycogen storage occurs in premature newborns or newborns that had intrauterine growth retardation. [2] Increased glucose use [ edit ] Major causes of increased glucose use in a newborn include hyperthermia, polycythemia, sepsis, and growth hormone deficiency. [2] Decreased gluconeogenesis [ edit ] Two major issues that cause decreased gluconogeneis are inborn errors of metabolism and adrenal insufficiency. [2] Depeleted glycogen stores [ edit ] Most common causes of depleted glycogen stores are starvation and asphyxia-perinatal stress. [2] Mechanism [ edit ] There are many types of hypoglycemia, including transient and reoccurring. [1] Each is associated with different risk factors [1] and may have many underlying causes.
Sulfonic acids : Acamprosate Religion and alcohol Christian views on alcohol alcohol in the Bible Islam and alcohol History Bratt System Related Index of alcohol-related articles Austrian syndrome Ban on caffeinated alcoholic beverages Brief intervention Gateway drug effect Last call Mood disorder Non-alcoholic fatty liver disease Self-medication Spins Sober companion Sober living houses Sobering center Town drunk Category
Sulfonic acids : Acamprosate Religion and alcohol Christian views on alcohol alcohol in the Bible Islam and alcohol History Bratt System Related Index of alcohol-related articles Austrian syndrome Ban on caffeinated alcoholic beverages Brief intervention Gateway drug effect Last call Mood disorder Non-alcoholic fatty liver disease Self-medication Spins Sober companion Sober living houses Sobering center Town drunk Category v t e Turkey topics History Overview Renaissance (1400-1500) Conquest of Constantinople Early modern period (1500-1750) Sultanate of Women Köprülü era Tulip era Late modern period (1750-1923) Tanzimat Ottoman Reform Edict of 1856 First Constitutional Era Second Constitutional Era Partition Contemporary period (1923-present) War of Independence One-party period Multi-party period By topic Constitutional Economic Empire Foreign relations 1814–1919 Military Society and its environment Overview Climate Boundaries Geology Landform regions By topic Education Language reform Health and welfare Individual, family and gender relations LGBT rights Marriage Status of women Population Population distribution and settlement in Turkey Migration Government policies Religious life Economy Overview Growth of the economy Development planning Economic development Foreign economic relations Foreign trade Regional economic integration By sector Agriculture Industry Construction Energy Mineral resources Services Banking and Finance Transportation and Telecommunications Tourism Government and politics The constitutional system Provisions Electoral system Government Parliament President Council of Ministers Prime minister Judiciary Constitutional Court Court of Cassation Political Dynamics Political parties Mass media Newspapers and periodicals Radio and television Foreign relations National security in Turkey External security concerns Middle Eastern conflicts Syria Iran Greece and Cyprus Military Participation in NATO Defense spending Sources and quality of personnel Education and training Air Force Navy Uniforms, ranks, and insignia Domestic arms industry Internal security concerns Kurdish separatism Armenian terrorism Islamists Police system National Police Gendarmerie Intelligence Services Individual rights Culture Architecture Ottoman architecture Art Cinema Cuisine wine Dance Festivals Folklore Languages Turkish Literature Media Newspapers Radio stations TV Music Names Theater Category Portal WikiProject