-
10q26 Deletion Syndrome
Medlineplus
10q26 deletion syndrome is a condition that results from the loss (deletion) of a small piece of chromosome 10 in each cell. ... The signs and symptoms of 10q26 deletion syndrome vary widely, even among affected members of the same family. ... A range of facial features is seen in people with 10q26 deletion syndrome, but not all affected individuals have these features. ... Frequency 10q26 deletion syndrome is thought to be a rare condition; at least 100 cases have been described in the scientific literature. ... The signs and symptoms of 10q26 deletion syndrome are probably related to the loss of one or more genes in the deleted region.
-
8q21.11 Microdeletion Syndrome
Orphanet
8q21.11 microdeletion syndrome encompasses heterozygous overlapping microdeletions on chromosome 8q21.11 resulting in intellectual disability, facial dysmorphism comprising a round face, ptosis, short philtrum, Cupid's bow and prominent low-set ears, nasal speech and mild finger and toe anomalies. Epidemiology The prevalence is unknown but 8q21.11 microdeletion syndrome is rare. To date, 13 cases, of which 5 from the same family, have been clinically and molecularly characterized without a notable gender discrepancy. ... Differential diagnosis Differential diagnosis includes Schilbach-Rott syndrome, auriculo-condylar (question mark ear) syndrome, Frydman syndrome, Kabuki syndrome (see these terms). The syndrome also bares resemblance with distal 22q11.2 microdeletion syndrome and 10p13 microdeletion syndrome. The entity should not be confused with the 8q22.1 microdeletion (see this term) found in patients with Nablus mask-like facial syndrome. Antenatal diagnosis Antenatal diagnosis of 8q21.11 microdeletion is possible by amniocentesis or chorionic villus sampling and cytogenetic analysis.
-
Anterior Compartment Syndrome
Wikipedia
Anterior compartment syndrome of the lower leg Specialty Rheumatology A compartment syndrome is an increased pressure within a muscular compartment [1] that compromises the circulation to the muscles. ... Sensitivity to passive stretch and active contraction are common, and tend to increase the symptoms. [ citation needed ] Pathology [ edit ] A compartment space is anatomically determined by an unyielding fascial (and osseous ) enclosure of the muscles. The anterior compartment syndrome of the lower leg (often referred to simply as anterior compartment syndrome), can affect any and all four muscles of that compartment: tibialis anterior , extensor hallucis longus , extensor digitorum longus , and peroneus tertius . [ citation needed ] This term is often mistakenly used to describe various related/proximal conditions, including Anterior Shin Splints . It is important to distinguish between the two, as shin splints rarely causes serious health problems, while Anterior Compartment Syndrome can lead to irreversible damage. [ citation needed ] The true compartment syndrome arises due to increased pressure within the unyielding anterior compartment of the leg . ... The process can begin with swelling of the tibialis anterior, extensor hallucis longus, extensor digitorum longus, and/or the peroneus tertius muscles in response to strong eccentric contractions sufficient to produce postexercise soreness. [ citation needed ] Diagnosis [ edit ] If these symptoms are observed/experienced it is important to contact a physician specializing in sports medicine ( MD / DO ), a doctor of podiatric medicine (DPM), or other qualified health care professional immediately so as to get the appropriate advice/treatment before serious damage occurs. [ citation needed ] The 5 Ps of Anterior Compartment Syndrome: Pain Pallor Paresthesia Pulselessness Paralysis (If not treated) Treatment [ edit ] The only option to treat acute compartment syndrome is surgery. ... Options to treat chronic compartment syndrome include physiotherapy, shoe inserts, and anti-inflammatory medications. [ citation needed ] References [ edit ] ^ Janet G.
-
Meier-Gorlin Syndrome
Medlineplus
Meier-Gorlin syndrome is a condition primarily characterized by short stature. ... Most people with Meier-Gorlin syndrome have distinctive facial features. ... Additional features of Meier-Gorlin syndrome can include difficulty feeding and a lung condition known as pulmonary emphysema or other breathing problems. Frequency Meier-Gorlin syndrome is a rare condition; however, its prevalence is unknown. Causes Meier-Gorlin syndrome can be caused by mutations in one of several genes.
-
Neuroleptic-Induced Deficit Syndrome
Wikipedia
Neuroleptic-induced deficit syndrome ( NIDS ) is a psychopathological syndrome that develops in some patients who take high doses of an antipsychotic for an extended time. [1] It is most often caused by high-potency typical antipsychotics , but can also be caused by high doses of many atypicals , especially those closer in profile to typical ones (that have higher D 2 dopamine receptor affinity and relatively low 5-HT 2 serotonin receptor binding affinity), like paliperidone and amisulpride . [2] Symptoms [ edit ] Neuroleptic induced deficit syndrome is principally characterized by the same symptoms that constitute the negative symptoms of schizophrenia : emotional blunting , apathy , hypobulia , anhedonia , indifference, difficulty or total inability in thinking, difficulty or total inability in concentrating, lack of initiative , attention deficits, and desocialization. [2] This can easily lead to misdiagnosis and mistreatment. Instead of decreasing the antipsychotic, the doctor may increase their dose to try to "improve" what they perceive to be negative symptoms of schizophrenia, rather than antipsychotic side effects. [ citation needed ] The concept of neuroleptic induced deficit syndrome was initially presented for schizophrenia , and it has rarely been associated in other mental disorders. [2] In recent years, atypical neuroleptics are being more often managed to patients with bipolar disorder , so some studies about neuroleptic-induced deficit syndrome in bipolar disorder patients now available. [2] There are significant difficulties in the differential diagnosis of primary negative symptoms and neuroleptic deficiency syndrome (secondary negative symptoms), as well as depression . [3] Case [ edit ] A Japanese man, who was being treated for schizophrenia, exhibited neuroleptics-induced deficit syndrome and obsessive–compulsive symptoms. [4] His symptoms were remarkably improved by quitting a course of antipsychotics followed by the introduction of the antidepressant fluvoxamine . [4] He had been misdiagnosed with schizophrenia, the real diagnosis was obsessive–compulsive disorder . [4] References [ edit ] ^ "Neuroleptic Induced Deficit Syndrome". ... PMID 7903967 . ^ a b c d Ueda S, Sakayori T, Omori A, Fukuta H, Kobayashi T, Ishizaka K, et al. (2016). "Neuroleptic-induced deficit syndrome in bipolar disorder with psychosis" . ... "How to distinguish between the neuroleptic-induced deficit syndrome, depression and disease-related negative symptoms in schizophrenia". ... 強迫性障害と抗精神病薬による欠陥症候群(NIDS)の合併例に抗精神病薬中止とSSRIが奏効した一例 [Case of obsessive-compulsive disorder associated with neuroleptics-induced deficit syndrome (NIDS): successfully treated by discontinuation of neuroleptics followed by SSRI.].
-
Zieve's Syndrome
Wikipedia
Zieve's syndrome Specialty Gastroenterology Zieve's syndrome is an acute metabolic condition that can occur during withdrawal from prolonged alcohol abuse. ... "Hemolysis in Acute Alcoholic Hepatitis: Zieve's Syndrome" . ACG Case Reports Journal . 2 (4): 250–251. doi : 10.14309/crj.2015.75 . ... "Alcohol-associated haemolysis in Zieve's syndrome: a clinical and laboratory study of five cases". ... "The significance of plasma phospholipids in Zieve syndrome". Blut . 21 (4): 210–226. ISSN 0006-5242 . ... "Jaundice, hyperlipemia and hemolytic anemia: a heretofore unrecognized syndrome associated with alcoholic fatty liver and cirrhosis".
-
Ichthyosis Follicularis With Alopecia And Photophobia Syndrome
Wikipedia
IFAP syndrome Ichthyosis follicularis with alopecia and photophobia syndrome is inherited via X-linked recessive manner(though other forms of inheritance have occurred) [1] Specialty Medical genetics IFAP syndrome is an extremely rare genetic syndrome. ... The gene or genes causing this disease are not known. [4] Diagnosis [ edit ] Diagnosis is based on appearance and family history. KID syndrome or keratosis follicularis spinulosa decalvans have some similar symptoms and must be eliminated. [5] Management [ edit ] Our son has IFAD syndrome. ... Regarding Ear infection - We mix a little vinegar with olive oil (or water) and apply two drops into the ear while he sleeps. [ citation needed ] See also [ edit ] Cicatricial alopecia List of cutaneous conditions References [ edit ] ^ "OMIM Entry - # 308205 - IFAP SYNDROME WITH OR WITHOUT BRESHECK SYNDROME" . omim.org . ... "Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome" . Orphanet Journal of Rare Diseases . 6 : 29. doi : 10.1186/1750-1172-6-29 . ... PMID 21600032 . ^ a b OMIM (Online Mendelian Inheritance in Man), Johns Hopkins University, Ichthyosis follicularis, atrichia and photophobia ^ Boente M del, Bibas-Bonet H, Coronel AM, Asial RA; Atrichia, ichthyosis, follicular hyperkeratosis, chronic candidiasis, keratitis, seizures, mental retardation and inguinal hernia: a severe manifestation of IFAP syndrome? , European Journal of Dermatology.

-
Amenorrhea-Galactorrhea Syndrome
Omim
The association of secondary amenorrhea and galactorrhea is generally thought to occur in 2 distinct syndromes: the Forbes-Albright syndrome, in which amenorrhea and galactorrhea are accompanied by a pituitary tumor, with or without prior pregnancy, and the Chiari-Frommel syndrome, in which amenorrhea and galactorrhea commence after pregnancy, without associated pituitary tumor. This distinction may be artificial (Rimoin and Schimke, 1971), because the pituitary adenoma may be too small to identify clinically and progression from the benign to the neoplastic syndrome has been documented (Young et al., 1967). ... Since the amenorrhea-galactorrhea syndrome has been described as a part of a multiple endocrine adenomatosis syndrome, it is not certain that the ailment in the mother and daughter reported by Linquette et al. (1967) represented a distinct entity.
-
Say Syndrome
Wikipedia
Say syndrome Other names Say-Barber-Hobbs syndrome Specialty Dermatology Say syndrome is a condition characterized by bilateral acromial dimples. [1] In an article published in Humangenetik, Say et al. (1975) described a 'new,' presumably autosomal dominant disorder characterized by cleft palate, short stature, microcephaly, large ears, and hand anomalies. [2] [3] See also [ edit ] Rud syndrome List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... ISBN 978-1-4160-2999-1 . ^ "OMIM Entry - 181180 - SAY SYNDROME" . omim.org . Retrieved 2020-08-05 . ^ Say, B.; Barber, D. ... G. (1975). "A new dominantly inherited syndrome of cleft palate" . Humangenetik . 26 (3): 267–269. doi : 10.1007/BF00281464 (inactive 2021-01-14).
-
Intersection Syndrome
Wikipedia
Unsourced or poorly sourced material may be challenged and removed . Find sources: "Intersection syndrome" – news · newspapers · books · scholar · JSTOR ( March 2019 ) Intersection syndrome Specialty Orthopedic Intersection syndrome is a painful condition that affects the lateral side of the forearm [1] when inflammation occurs at the intersection of the muscle bellies of the abductor pollicis longus and extensor pollicis brevis cross over the extensor carpi radialis longus and the extensor carpi radialis brevis . ... The mechanism of injury is usually repetitive resisted extension, as with rowing, weight lifting, or pulling. [ citation needed ] Intersection syndrome is often confused with another condition called DeQuervain's syndrome , which is an irritation of the thumb-sided set of tendons at the wrist, called the first dorsal compartment. References [ edit ] ^ "Intersection Syndrome: Practice Essentials, Problem, Epidemiology" .
-
Familial Hypocalciuric Hypercalcemia
Wikipedia
External links [ edit ] Classification D ICD - 10 : E83.5 OMIM : 145980 145981 600740 MeSH : D006934 DiseasesDB : 1326 External resources Orphanet : 405 v t e Electrolyte imbalances Sodium High Salt poisoning Low Hypotonic Isotonic Cerebral salt-wasting syndrome Potassium High Low Chloride High Low Calcium High Low Symptoms and signs Chvostek sign Trousseau sign Milk-alkali syndrome Disorders of calcium metabolism Calcinosis ( Calciphylaxis , Calcinosis cutis ) Calcification ( Metastatic calcification , Dystrophic calcification ) Familial hypocalciuric hypercalcemia Phosphate High Low Magnesium High Low v t e Cell surface receptor deficiencies G protein-coupled receptor (including hormone ) Class A TSHR ( Congenital hypothyroidism 1 ) LHCGR ( Luteinizing hormone insensitivity , Leydig cell hypoplasia , Male-limited precocious puberty ) FSHR ( Follicle-stimulating hormone insensitivity , XX gonadal dysgenesis ) GnRHR ( Gonadotropin-releasing hormone insensitivity ) EDNRB ( ABCD syndrome , Waardenburg syndrome 4a , Hirschsprung's disease 2 ) AVPR2 ( Nephrogenic diabetes insipidus 1 ) PTGER2 ( Aspirin-induced asthma ) Class B PTH1R ( Jansen's metaphyseal chondrodysplasia ) Class C CASR ( Familial hypocalciuric hypercalcemia ) Class F FZD4 ( Familial exudative vitreoretinopathy 1 ) Enzyme-linked receptor (including growth factor ) RTK ROR2 ( Robinow syndrome ) FGFR1 ( Pfeiffer syndrome , KAL2 Kallmann syndrome ) FGFR2 ( Apert syndrome , Antley–Bixler syndrome , Pfeiffer syndrome , Crouzon syndrome , Jackson–Weiss syndrome ) FGFR3 ( Achondroplasia , Hypochondroplasia , Thanatophoric dysplasia , Muenke syndrome ) INSR ( Donohue syndrome Rabson–Mendenhall syndrome ) NTRK1 ( Congenital insensitivity to pain with anhidrosis ) KIT ( KIT Piebaldism , Gastrointestinal stromal tumor ) STPK AMHR2 ( Persistent Müllerian duct syndrome II ) TGF beta receptors : Endoglin / Alk-1 / SMAD4 ( Hereditary hemorrhagic telangiectasia ) TGFBR1 / TGFBR2 ( Loeys–Dietz syndrome ) GC GUCY2D ( Leber's congenital amaurosis 1 ) JAK-STAT Type I cytokine receptor : GH ( Laron syndrome ) CSF2RA ( Surfactant metabolism dysfunction 4 ) MPL ( Congenital amegakaryocytic thrombocytopenia ) TNF receptor TNFRSF1A ( TNF receptor associated periodic syndrome ) TNFRSF13B ( Selective immunoglobulin A deficiency 2 ) TNFRSF5 ( Hyper-IgM syndrome type 3 ) TNFRSF13C ( CVID4 ) TNFRSF13B ( CVID2 ) TNFRSF6 ( Autoimmune lymphoproliferative syndrome 1A ) Lipid receptor LRP : LRP2 ( Donnai–Barrow syndrome ) LRP4 ( Cenani–Lenz syndactylism ) LRP5 ( Worth syndrome , Familial exudative vitreoretinopathy 4 , Osteopetrosis 1 ) LDLR ( LDLR Familial hypercholesterolemia ) Other/ungrouped Immunoglobulin superfamily : AGM3, 6 Integrin : LAD1 Glanzmann's thrombasthenia Junctional epidermolysis bullosa with pyloric atresia EDAR ( EDAR hypohidrotic ectodermal dysplasia ) PTCH1 ( Nevoid basal-cell carcinoma syndrome ) BMPR1A ( BMPR1A juvenile polyposis syndrome ) IL2RG ( X-linked severe combined immunodeficiency ) See also cell surface receptors v t e Congenital endocrine disorders Pituitary Congenital hypopituitarism Thyroid Thyroid disease Persistent thyroglossal duct Thyroglossal cyst Congenital hypothyroidism Thyroid dysgenesis Thyroid dyshormonogenesis Pendred syndrome Parathyroid Congenital absence of parathyroid Adrenal Absent adrenal gland
-
Thoracic Outlet Syndrome
Mayo_clinic
Sometimes doctors don't know the cause of thoracic outlet syndrome. Treatment for thoracic outlet syndrome usually involves physical therapy and pain relief measures. ... Symptoms There are three general types of thoracic outlet syndrome: Neurogenic (neurologic) thoracic outlet syndrome. ... Arterial thoracic outlet syndrome. This is the least common type of TOS . ... Thoracic outlet syndrome symptoms can vary depending on the type. ... It's often the first imaging test used to help diagnose thoracic outlet syndrome. Doctors may use this test to see if you have vascular thoracic outlet syndrome or other vascular problems.

-
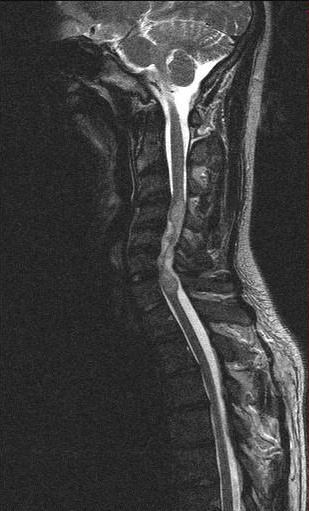
Froin's Syndrome
Wikipedia
Froin's syndrome Ependymoma of the cervical spine , completely obscurating the spinal canal Froin's syndrome – coexistence of xanthochromia , high protein level and marked coagulation of cerebrospinal fluid (CSF). ... It often shows the prolongation of T1 and T2 signal in CSF caudal to a level of block. [2] This phenomenon is named after Georges Froin (1874–1932), a French physician who first described it. [3] [4] References [ edit ] ^ Govindarajan, R; Khan, T (August 2012). "Froin's syndrome: an uncommon mimicker of Guillain–Barre syndrome" . ... PMID 22447409 . ^ Mirza, S; Adams, WM; Corkhill, RA (May 2008). "Froin's syndrome revisited, 100 years on. Pseudo-Froin's syndrome on MRI". Clinical Radiology . 63 (5): 600–4. doi : 10.1016/j.crad.2007.07.027 . PMID 18374725 . ^ Froin's syndrome at Who Named It? ^ Froin G. Inflammations méningées avec chromatique, fibrineuse et cytologique du liquide céphalo-rachidien .
-
Boucher-Neuhäuser Syndrome
Medlineplus
Boucher-Neuhäuser syndrome is a rare disorder that affects movement, vision, and sexual development. ... The third characteristic feature of Boucher-Neuhäuser syndrome is eye abnormalities, most commonly chorioretinal dystrophy. ... The key features of Boucher-Neuhäuser syndrome can begin anytime from infancy to adulthood, although at least one feature usually occurs by adolescence. ... Frequency Boucher-Neuhäuser syndrome is a rare condition. Its prevalence is unknown. Causes Most cases of Boucher-Neuhäuser syndrome are caused by mutations in the PNPLA6 gene.
-
Megalencephaly-Polymicrogyria-Polydactyly-Hydrocephalus Syndrome
Medlineplus
Megalencephaly-polymicrogyria-polydactyly-hydrocephalus (MPPH) syndrome is a rare disorder that primarily affects the development of the brain. ... People with MPPH syndrome have delayed development and intellectual disability that ranges from mild to severe. ... About half of people with MPPH syndrome have an extra finger or toe on one or more of their hands or feet (polydactyly). ... The brain abnormalities characteristic of MPPH syndrome are also found in a closely related condition called megalencephaly-capillary malformation syndrome (MCAP). However, MCAP includes abnormalities of small blood vessels in the skin (capillary malformations) and several other features that are not usually part of MPPH syndrome. Frequency MPPH syndrome appears to be a rare disease.
-
Glut1 Deficiency Syndrome
Medlineplus
GLUT1 deficiency syndrome is a disorder affecting the nervous system that can have a variety of neurological signs and symptoms. ... People with this form of GLUT1 deficiency syndrome may have developmental delay or intellectual disability. ... About 10 percent of individuals with GLUT1 deficiency syndrome have a form of the disorder often known as non-epileptic GLUT1 deficiency syndrome, which is usually less severe than the common form. ... Frequency GLUT1 deficiency syndrome is a rare disorder. Approximately 500 cases have been reported worldwide since the disorder was first identified in 1991. ... About 90 percent of cases of GLUT1 deficiency syndrome result from new mutations in the gene.
-
Synostosis
Wikipedia
Pain is generally not a problem, unless radial head dislocation should occur. [1] [2] Most examples of radioulnar synostosis are isolated (non-syndromic). Syndromes that may be accompanied by radioulnar synostosis include X chromosome polyploidy (e.g., XXXY ) and other chromosome disorders (e.g., 4p- syndrome , Williams syndrome ), acrofacial dysostosis , Antley–Bixler syndrome , genitopatellar syndrome , Greig cephalopolysyndactyly syndrome , hereditary multiple osteochondromas ( hereditary multiple exostoses ), limb-body wall complex , and Nievergelt syndrome . ... Retrieved 24 April 2015 . ^ Craniosynostosis External links [ edit ] Synostosis at the US National Library of Medicine Medical Subject Headings (MeSH) v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatumFGFR3, TWIST1, TCF12, GDF6, NOG, FGFR2, HOXA11, MSX2, PBX1, CUP2Q35, CYP26B1, ZIC1, TGFB2, POR, RUNX2, NELL1, EFNA4, LRPAP1, IGF1R, IGF1, FLNB, FGFR1, FGF9, EFNB1, SMAD6

-
Lacrimo-Auriculo-Dento-Digital Syndrome
Medlineplus
Lacrimo-auriculo-dento-digital (LADD) syndrome is a genetic disorder that mainly affects the eyes, ears, mouth, and hands. LADD syndrome is characterized by defects in the tear-producing lacrimal system (lacrimo-), ear problems (auriculo-), dental abnormalities (dento-), and deformities of the fingers (digital). ... People with LADD syndrome may have underdeveloped or absent salivary glands , which impairs saliva production. ... Hand deformities are also a frequent feature of LADD syndrome. Affected individuals may have abnormally small or missing thumbs . ... It can be shorter than normal with abnormal wrist and elbow joint development that limits movement. People with LADD syndrome may also experience other signs and symptoms.
-
Bruck Syndrome 2
Omim
A number sign (#) is used with this entry because Bruck syndrome-2 (BRKS2) is caused by homozygous mutation in the PLOD2 gene (601865), which encodes telopeptide lysyl hydroxylase, on chromosome 3q24. For a phenotypic description and a discussion of genetic heterogeneity of Bruck syndrome, see Bruck syndrome-1 (259450). Clinical Features Ha-Vinh et al. (2004) described a child with Bruck syndrome who was the offspring of healthy nonconsanguineous Turkish parents. ... Van der Slot et al. (2003) stated that they were unaware of any phenotypic differences between Bruck syndromes 1 and 2. Biochemical Features Bank et al. (1999) reported that the molecular defect underlying Bruck syndrome is a deficiency of bone-specific telopeptide lysyl hydroxylase, which results in aberrant crosslinking of bone collagen. ... In contrast to bone, cartilage and ligament showed unaltered telopeptide hydroxylation in Bruck syndrome, as evidenced by normal patterns of crosslinking.
-
Stüve-Wiedemann Syndrome
Medlineplus
Another condition once known as Schwartz-Jampel syndrome type 2 is now considered to be part of Stüve-Wiedemann syndrome. Researchers have recommended that the designation Schwartz-Jampel syndrome type 2 no longer be used. Frequency Stüve-Wiedemann syndrome is a rare condition that has been found worldwide. Its prevalence is unknown. Causes Stüve-Wiedemann syndrome is usually caused by mutations in the LIFR gene. ... Most LIFR gene mutations that cause Stüve-Wiedemann syndrome prevent production of any LIFR protein. ... A small number of people with Stüve-Wiedemann syndrome do not have an identified mutation in the LIFR gene.



