Gradenigo's syndrome Apicitis petrosa as seen in computed tomography Specialty Otorhinolaryngology Gradenigo's syndrome , also called Gradenigo-Lannois syndrome , [1] [2] is a complication of otitis media and mastoiditis involving the apex of the petrous temporal bone . ... The syndrome is classically caused by the spread of an infection into the petrous apex of the temporal bone . ... "[Some cases of Gradenigo-Lannois syndrome]". Journal de Médecine de Lyon . 47 (96): 537–47. ... "[Cranial osteitis: a report on four cases, including a Gradenigo-Lannois syndrome (author's transl)]". Journal de Radiologie . 61 (11): 677–81. ... PMID 21490711 . ^ Motamed M, Kalan A (September 2000). "Gradenigo's syndrome" . Postgraduate Medical Journal . 76 (899): 559–60. doi : 10.1136/pmj.76.899.559 .
Thiamine-responsive megaloblastic anemia syndrome is a rare condition characterized by hearing loss, diabetes, and a blood disorder called megaloblastic anemia. ... Individuals with thiamine-responsive megaloblastic anemia syndrome begin to show symptoms of megaloblastic anemia between infancy and adolescence. This syndrome is called "thiamine-responsive" because the anemia can be treated with high doses of vitamin B1 (thiamine). ... People with thiamine-responsive megaloblastic anemia syndrome usually require insulin to treat their diabetes . ... Heart and blood vessel (cardiovascular) problems such as heart rhythm abnormalities and heart defects have also been reported in some people with this syndrome. Frequency Thiamine-responsive megaloblastic anemia syndrome has been reported in approximately 30 families worldwide.
Thiamine-responsive megaloblastic anemia syndrome (TRMA) is characterized by megaloblastic anemia, progressive sensorineural hearing loss, and diabetes mellitus. ... One case report suggests "thiamine-responsive myelodysplasia" as an alternative name for the syndrome [Bazarbachi et al 1998]. Prevalence Approximately 50 pedigrees are known. ... Among acquired anemias, this combination is most suggestive of myelodysplastic syndromes in which megaloblastosis and sideroblasts are often noted. ... Notably missing in Wolfram syndrome are megaloblastic anemia and thiamine responsiveness. Wolfram syndrome is caused by pathogenic variants in WFS1 .
Thiamine-responsive megaloblastic anemia (TRMA) is characterized by a triad of megaloblastic anemia, non-type I diabetes mellitus, and sensorineural deafness. Epidemiology TRMA syndrome has been reported in less than 80 cases worldwide. ... The variable phenotypic presentation of TRMA syndrome may cause a significant delay between the onset of symptoms and an accurate diagnosis. ... Differential diagnosis Differential diagnosis includes Wolfram syndrome, mitochondrial disorders such as Kearns-Sayre syndrome and Pearson syndrome (see these terms), as well as dietary vitamin B12 or folate deficiency.
A number sign (#) is used with this entry because of evidence that thiamine-responsive megaloblastic anemia syndrome (TRMA), also known as thiamine metabolism dysfunction syndrome-1 (THMD1), is caused by homozygous mutation in the SLC19A2 (603941) gene, which encodes a thiamine transporter protein, on chromosome 1q24. Description Thiamine-responsive megaloblastic anemia syndrome comprises megaloblastic anemia, diabetes mellitus, and sensorineural deafness. ... The parents were first cousins and were partially deaf. The syndrome was further delineated and autosomal recessive inheritance corroborated by Haworth et al. (1982), who described affected Pakistani brother and sister. ... The abnormalities in the thiamine-responsive anemia syndrome are consistent with the picture of thiamine-deficient beriberi in childhood (Burgess, 1958). ... However, a later study by Neufeld et al. (1997) determined that the patients reported by Borgna-Pignatti et al. (1989) in fact had thiamine-responsive megaloblastic anemia syndrome, with linkage to chromosome 1q.
Thiamine-responsive megaloblastic anemia syndrome is a very rare condition characterized by hearing loss, diabetes , and a blood disorder called megaloblastic anemia . Affected individuals begin to show symptoms of this condition between infancy and adolescence. This syndrome is called "thiamine-responsive" because the anemia can be treated with high doses of vitamin B1 (thiamine) .
Thiamine responsive megaloblastic anemia syndrome Specialty Medical genetics Complications Diabetes mellitus , anemia , hearing loss Causes SLC19A2 gene mutation [1] Treatment Thiamine Thiamine responsive megaloblastic anemia syndrome (also known as Rogers Syndrome) is a very rare autosomal recessive genetic disorder affecting a thiamine transporter, which is characterized by megaloblastic anemia , diabetes mellitus , and hearing loss . ... Contents 1 Signs and symptoms 2 Treatment 3 Genetics 4 History 5 References 6 External links Signs and symptoms [ edit ] In most cases (80-99%), people with this condition experience poor appetite (anorexia) , diarrhea , headache, and lethargy. [1] Thiamine responsive megaloblastic anemia syndrome is associated with progressive sensorineural hearing loss . ... References [ edit ] ^ a b c "Thiamine responsive megaloblastic anemia syndrome" . Genetic and Rare Diseases Information Center . ... Indian Pediatrics . 46 . ^ "Thiamine-responsive megaloblastic anemia syndrome" . Genetics Home Reference . US National Library of Medicine.
Nutcracker syndrome Other names Nutcracker phenomenon, renal vein entrapment syndrome, mesoaortic compression of the left renal vein The nutcracker syndrome results from compression of the left renal vein between the aorta and the superior mesenteric artery . ... The legs of this nutcracker, with some imagination, could represent the superior mesenteric artery and abdominal aorta in nutcracker syndrome. Play media Summary video explaining signs and symptoms as well as etiology of nutcracker syndrome. ... PMID 20511485 . ^ Sugimoto I, Takashi O, Ishibashi H, Takeuchi N, Nagata Y, Honda Y (2001). "Left Renal Vein Entrapment Syndrome (Nutcracker Syndrome) treated with Left Renal Vein Transposition" . ... S2CID 35732843 . ^ a b Hilgard P, Oberholzer K, Meyer zum Büschenfelde KH, Hohenfellner R, Gerken G (July 1998). "[The "nutcracker syndrome" of the renal vein (superior mesenteric artery syndrome) as the cause of gastrointestinal complaints]". ... "What Each Clinical Anatomist Has to Know about Left Renal Vein Entrapment Syndrome (Nutcracker Syndrome): A Review of the Most Important Findings" .
Renal nutcracker syndrome (NCS) is a condition that occurs when the left renal vein (the vein that carries blood purified by the left kidney) becomes compressed.
A rare, syndromic renal disease characterized by the entrapment of left renal vein (LRV) between the superior mesenteric artery (SMA) and the abdominal aorta, resulting in increased luminal pressure, renal hilar varices, hematuria and, at the microscopic level, rupture of thin-walled veins into the collecting system in renal fornices. ... Clinical description Patients with renal nutcracker syndrome (NCS) are usually asthenic, tall and thin. ... Gynecological symptoms resemble pelvic congestion syndrome and include symptoms of dysmenorrhea, dyspareunia, post-coital ache, lower abdominal pain, dysuria, pelvic/vulvar/gluteal/gonadal or thigh varices, and emotional disturbances. Three types of renal nutcracker syndrome have been defined, according to the site of LVR compression: anterior nutcracker syndrome, posterior nutcracker syndrome and combined nutcracker syndrome.
Genetic Heterogeneity of Robinow Syndrome Autosomal recessive Robinow syndrome-2 (RRS2; 618529) is caused by mutation in the NXN gene (612895) on chromosome 17p13. ... He suggested that the cases so designated represent a recessive form of Robinow syndrome. Glaser et al. (1989) described Robinow syndrome in a child of a consanguineous Turkish couple. ... Nazer et al. (1990) reported 2 children, offspring of first-cousin Saudi parents, with the 'fetal face syndrome.' Both of the children also had the Crigler-Najjar syndrome, as did 2 previously born sibs who did not have the fetal face syndrome. ... Vaginal atresia with cervical agenesis had apparently not been observed previously in Robinow syndrome. This was thought to represent a recessive form of Robinow syndrome. ... Beiraghi et al. (2011) compared the craniofacial and intraoral phenotypes of 9 patients with dominant Robinow syndrome to 3 patients with recessive Robinow syndrome.
Autosomal recessive Robinow syndrome (RRS) is the less common type of Robinow syndrome (RS, see this term) characterized by short-limb dwarfism, costovertebral segmentation defects and abnormalities of the head, face and external genitalia. ... Rib fusions are considered to be characteristic of the autosomal recessive form. Etiology The syndrome is caused by mutations in the ROR2 gene (9q22).
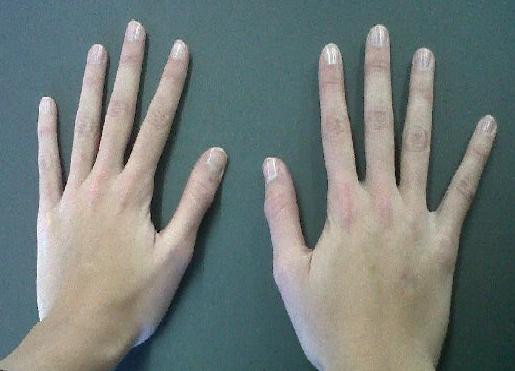
Glanzmann thrombasthenia is a bleeding disorder that is characterized by prolonged or spontaneous bleeding starting from birth. People with Glanzmann thrombasthenia tend to bruise easily, have frequent nosebleeds (epistaxis), and may bleed from the gums. They may also develop red or purple spots on the skin caused by bleeding underneath the skin (petechiae) or swelling caused by bleeding within tissues (hematoma). Glanzmann thrombasthenia can also cause prolonged bleeding following injury, trauma, or surgery (including dental work). Women with this condition can have prolonged and sometimes abnormally heavy menstrual bleeding.
Montgomery et al. (1983) demonstrated that an assay using monoclonal antibodies raised in the mouse can recognize the deficiency of glycoprotein Ib in the Bernard-Soulier syndrome (BSS; 231200) and of the glycoprotein IIb/IIIa in Glanzmann thrombasthenia.
Glanzmann thrombasthenia (GT) is a bleeding syndrome characterized by spontaneous mucocutaneous bleeding and an exaggerated response to trauma due to a constitutional thrombocytopenia.
Glanzmann thrombasthenia (GT) is a rare inherited blood clotting disorder that is present at birth. It is characterized by the impaired function of specialized blood cells, called platelets, that are essential for proper blood clotting. Signs and symptoms vary greatly from person to person. Symptoms usually include abnormal bleeding, which can be severe. Other symptoms may include easy bruising, nose bleeds , bleeding from the gums, and/or heavy menstrual bleeding. Rarely, internal bleeding and blood in the urine ( hematuria ) can occur.
Fragile XE syndrome is a genetic disorder that impairs thinking ability and cognitive functioning. ... Females are rarely diagnosed with fragile XE syndrome, likely because the signs and symptoms are so mild that the individuals function normally. ... Unlike some other forms of intellectual disability, cognitive functioning remains steady and does not decline with age in fragile XE syndrome. Frequency Fragile XE syndrome is estimated to affect 1 in 25,000 to 100,000 newborn males. ... However, in people with fragile XE syndrome, the CCG segment is repeated more than 200 times, which makes this region of the gene unstable. ... Learn more about the gene associated with Fragile XE syndrome AFF2 Inheritance Pattern Fragile XE syndrome is inherited in an X-linked pattern.
Clinical Features Knight et al. (1996) provided clinical details of 1 FRAXE male identified through a screening study as well as 3 other FRAXE individuals identified through previous referrals for fragile X syndrome testing. The first male had shown developmental delay and microcephaly. At the age of 10 years he showed short stature and an 'engaging' personality, which suggested William syndrome. No abnormality at the elastin locus (130160) characteristic of William syndrome was found, however. ... However, several families originally described as having the fragile X syndrome on the basis of mental impairment and cytogenetic analysis were shown by fluorescence in situ hybridization (FISH) to express FRAXE. ... Knight et al. (1996) described the results of a UK survey designed to assess the frequency of FRAXE in a population of individuals referred for fragile X syndrome testing and found to be negative for expansion events at the FRAXA locus.
Fragile XE syndrome (FRAXE) is a genetic condition associated with mild to borderline intellectual disabilities with physical features differing from person to person. ... The repeating nucleotides in FRAXE syndrome are CCG. When the number of CCG repeats is over 200, people typically have the signs and symptoms seen in FRAXE.
A rare X-linked syndromic intellectual disability characterized by a variable clinical picture including developmental delay, mild to moderate intellectual disability, learning difficulties, communication deficits, and behavioral problems (such as aggression, attention deficit, hyperactivity, and autistic features).
Gorlin-Chaudhry-Moss syndrome is a condition that affects many parts of the body. ... Many people with Gorlin-Chaudhry-Moss syndrome have a lack of fatty tissue under the skin (lipodystrophy). ... People with Gorlin-Chaudhry-Moss syndrome also have shortened bones at the ends of the fingers and toes (short distal phalanges). ... Frequency Gorlin-Chaudhry-Moss syndrome is an extremely rare condition. ... Learn more about the gene associated with Gorlin-Chaudhry-Moss syndrome SLC25A24 Inheritance Pattern In individuals with an SLC25A24 gene mutation, Gorlin-Chaudhry-Moss syndrome is inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Progeroid syndrome, Petty type is a rare premature aging syndrome characterized by pre-and postnatal growth retardation, a congenital premature-aged appearance with distinctive craniofacial dysmorphism (wide calvaria with large open anterior fontanel and wide metopic suture, broad forehead, small face, micrognathia), markedly diminished subcutaneous fat, cutis laxa and wrinkled skin, without delay in psychomotor development.
A number sign (#) is used with this entry because of evidence that Fontaine progeroid syndrome (FPS) is caused by heterozygous mutation in the SLC25A24 gene (608744) on chromosome 1p36. ... Nomenclature Fontaine progeroid syndrome and Gorlin-Chaudhry-Moss syndrome (GCMS) were originally thought to be separate disorders. ... Clinical Features Petty et al. (1990) described what they considered to be a newly recognized form of congenital progeroid syndrome in a 5-year-old girl. They suggested that the 46-year-old woman reported by Wiedemann (1979) had the same syndrome. ... Despite some overlap in clinical manifestations, Faivre et al. (1999) also thought it unlikely that their infant had WRS, also known as neonatal progeroid syndrome. Rodriguez and Perez-Alonso (1999) defended the 'diagnosis of progeria syndrome [as] the only one possible.' ... The same sisters were reported by Feinberg (1960) as instances of the Weill-Marchesani syndrome (277600), which was clearly an incorrect diagnosis.
Gorlin-Chaudhry-Moss (GCM) syndrome is a multiple congenital anomaly syndrome characterized by craniofacial dysostosis, facial dysmorphism, conductive hearing loss, generalized hypertrichosis, and extremity, ocular and dental anomalies. ... Other additional features that may be observed include congenital laryngomalacia and heart disease (patent arterial duct) (see these terms). Progeroid syndrome, Petty type and Saethre-Chotzen syndrome (see these terms) have overlapping features with GCM syndrome and should be considered in the differential diagnosis.
Individuals experiencing solipsism syndrome feel that reality is not 'real' in the sense of being external to their own minds. The syndrome is characterized by feelings of loneliness, detachment and indifference to the outside world. ... Solipsism syndrome is distinct from solipsism , which is not a psychological state but rather a philosophical position, namely that nothing exists or can be known to exist outside of one's own mind; advocates of this philosophy do not necessarily suffer from solipsism syndrome, and sufferers do not necessarily subscribe to solipsism as a school of intellectual thought. ... Psychologists have noted how astronauts and cosmonauts exhibit symptoms of 'Solipsism Syndrome' - a mental condition ^ March, Scott F. (1984). ... Help for Solipsism Syndrome sufferers Dissociation from Living with Schizoaffective Disorder - A personal account of Solipsism Disorder
It is a common feature of several kinds of hereditary disorders which affect connective tissue , such as Marfan syndrome [2] and homocystinuria . Contents 1 In fiction 2 See also 3 References In fiction [ edit ] The condition is mentioned in the Rizzoli & Isles episode Boston Strangler Redux; Maura Isles ( Sasha Alexander ) is on a date with a man whom she diagnoses as having Marfan Syndrome, which she says "explains the dolichostenomelia."
Unsourced material may be challenged and removed. Find sources: "Wunderlich syndrome" – news · newspapers · books · scholar · JSTOR ( December 2018 ) ( Learn how and when to remove this template message ) Wunderlich syndrome Other names Double uterus-hemivagina-renal agenesis syndrome [1] This condition is inherited in an autosomal dominant manner Specialty Nephrology Wunderlich syndrome can refer to one of several conditions . One condition called Wunderlich syndrome is spontaneous, nontraumatic kidney bleeding confined to the subcapsular and perirenal space. It may be the first manifestation of a renal angiomyolipoma (AML), or the rupture of a renal artery or intraparenchymal aneurysm . [2] The renal condition should not be confused with other conditions which are Mullerian duct anomalies, such as Herlyn-Werner-Wunderlich syndrome. Some sources refer to double uters-hemivagina-renal agenesis as simply Wunderlich syndrome, but Herlyn-Werner-Wunderlich is a better term to distinguish the two. ... An ultrasound or CT scan can establish diagnosis, while lab tests may be inconclusive as changes of hematocrit or hemoglobin are not specific to the syndrome, while hematuria is not always present. ... Albi G, Campo L, Tagarro D (Sep 2002). "Wünderlich's syndrome: causes, diagnosis and radiological management".
Conditions that incriminate Kounis syndrome include bronchial asthma, Churg-Strauss syndrome, serum sickness, Scombroid syndrome, angioedema, hay fever, anaphylaxis (exercise induced or idiopathic), and Anisakiasis. ... Reactions to various foods that cause an allergic and inflammatory response can lead to acute coronary syndrome. [1] Signs and symptoms [ edit ] Allergic ACS is a syndrome involving 2 components. ... "Kounis syndrome: an update on epidemiology, pathogenesis, diagnosis and therapeutic management". ... S2CID 11386405 . ^ a b Abdelghany M, Subedi R, Shah S, Kozman H (April 2017). "Kounis syndrome: A review article on epidemiology, diagnostic findings, management and complications of allergic acute coronary syndrome". ... PMID 28153536 . ^ Memon S, Chhabra L, Masrur S, Parker MW (July 2015). "Allergic acute coronary syndrome (Kounis syndrome)" . Proceedings (Baylor University.
Mutations in the CHRNA1 (100690) and CHRND (100720) genes can also result in lethal multiple pterygium syndrome; mutations in these genes can also cause fast- or slow-channel congenital myasthenic syndromes (608930 and 601462, respectively). ... Van Regemorter et al. (1984) documented the lethal multiple pterygium syndrome in 2 spontaneously aborted fetuses from first-cousin parents of Moroccan origin. ... In brother and sister, Robinson et al. (1987) described a lethal type of multiple pterygium syndrome in which malignant hyperthermia was a major complication. ... The only family with reasonably clear X-linked prenatal lethal multiple pterygium syndrome was that reported by Tolmie et al. (1987). Molecular Genetics Hoffmann et al. (2006) and Morgan et al. (2006) found mutations in the CHRNG gene (e.g., 100730.0002) causing the lethal form of multiple pterygium syndrome. Michalk et al. (2008) found mutations in the CHRNA1 (e.g., 100690.0013) and CHRND (e.g., 100720.0005) genes that caused lethal multiple pterygium syndrome.
A rare genetic multiple pterygium syndrome characterized by intrauterine growth retardation, fetal akinesia, multiple joint contractures causing severe arthrogryposis and pterygia (webbing) across multiple joints. ... Clinical description Lethal multiple pterygium syndrome (LMPS) is characterized by growth deficiency of prenatal onset, pterygia present in multiple areas (chin to sternum, cervical, axillary, humero-ulnar, crural, popliteal and the ankles) and flexion contractures giving rise to severe arthrogryposis. ... These may include fetal akinesia deformation sequence (FADS), Bartsocas-Papas syndrome, Escobar variant multiple pterygium syndrome, arthrogryposis multiplex congenita, and maternal myasthenia gravis, as well as trisomy 18, severe neural tube defects, caudal regression sequence and vertebral anomalies, limb body wall complex, fetal neck masses, fetal hypoxia, constriction rings syndrome and fetal constraint.
Freeman-Sheldon syndrome is a condition that primarily affects the face, hands, and feet. ... Frequency Freeman-Sheldon syndrome is a rare disorder; its exact prevalence is unknown. Causes Freeman-Sheldon syndrome may be caused by mutations in the MYH3 gene. ... Some people with Freeman-Sheldon syndrome do not have mutations in the MYH3 gene. ... Learn more about the gene associated with Freeman-Sheldon syndrome MYH3 Inheritance Pattern Freeman-Sheldon syndrome can have different inheritance patterns.
Description Whistling face syndrome is characterized by an atypical facial appearance with anomalies of the hands and feet. ... Clinical Features The whistling face syndrome was first described as craniocarpotarsal dystrophy by Freeman and Sheldon (1938). Burian (1963) rediscovered the entity and called it the 'whistling face syndrome.' Zampino et al. (1996) described a sporadic case of the whistling face syndrome in a boy who also had severe hypertonicity, swallowing problems, and cerebellar and brainstem atrophy. The authors suggested that primary brain anomalies may explain many manifestations of the syndrome. They noted that it might be more appropriate to speak about the Freeman-Sheldon spectrum (193700) rather than syndrome because of the different pathogenetic mechanisms (muscular, skeletal, and neurologic), the wide range of clinical manifestations, and the genetic heterogeneity. ... Dallapiccola et al. (1989) described brother and sister with the whistling face syndrome who were born to unaffected parents.
A rare congenital, distal arthogryposis syndrome characterized by microstomia, whistling-face appearance, Chin with V- or H- shaped creased, and prominent nasolabial folds; most patients present club foot and congenital joint contractures of the hands and feet. ... Epidemiology Approximately 100 cases of Freeman-Sheldon syndrome (FSS) have been reported to date; no gender predominance is reported and the distribution is worldwide. ... Differential diagnosis Differential diagnosis mainly includes other distal arthrogryposis such as digitotalar dymorphism (which lacks craniofacial features), Sheldon-Hall syndrome (clinically less severe), Gordon syndrome, trismus-pseudocamptodactyly syndrome, and autosomal dominant multiple pterygium syndrome. Schwartz-Jampel syndrome and CLIFAHDD (congenital contractures of the limbs and face, hypotonia, and developmental delay) are also to be considered.
Mutations in this gene can also cause distal arthrogryposis type 2B (DA2B; 601680), also known as Sheldon-Hall syndrome. Description Freeman-Sheldon syndrome (FSS), or DA2A, is phenotypically similar to DA1. ... Fraser et al. (1970) reported the syndrome in father and son; dramatic pictures of their cases have been published (Gellis et al., 1970). ... The authors suggested that primary brain anomalies may explain many manifestations of the syndrome. They noted that it might be more appropriate to speak about the Freeman-Sheldon spectrum rather than syndrome because of the wide range of clinical manifestations. ... In the 41 non-FSS cases, the diagnoses included DA2B, or Freeman-Sheldon syndrome variant (601680) in 38, multiple pterygium syndrome (265000) in 1, and unknown in 2. ... Bamshad et al. (1996) presented a revised and extended classification of the distal arthrogryposes, and referred to Freeman-Sheldon syndrome as DA2 (later DA2A). DA2A is distinct from DAIIA (114300), which is also known as Gordon syndrome.
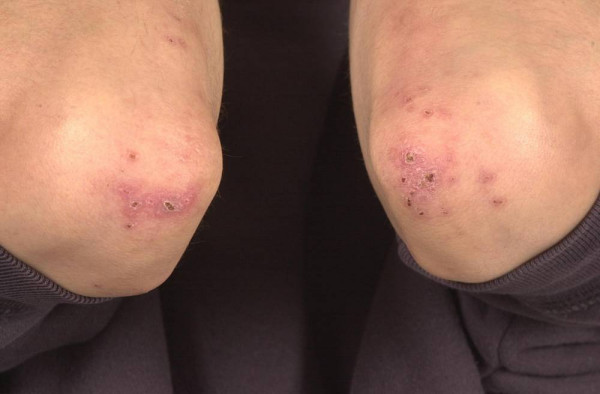
Bowel-associated dermatosis–arthritis syndrome Other names Bowel bypass syndrome and Intestinal bypass arthritis–dermatitis syndrome Pustules and crusts around the elbows in a patient with Crohn's disease and bowel-associated dermatosis-arthritis syndrome ( BADAS ) Specialty Dermatology Bowel-associated dermatosis–arthritis syndrome ( BADAS ), is a complication of jejunoileal bypass surgery consisting of flu-like symptoms ( fever , malaise ), multiple painful joints ( polyarthralgia ), muscle aches ( myalgia ) and skin changes. ... (March 1983). "Bowel-bypass syndrome without bowel bypass. Bowel-associated dermatosis-arthritis syndrome". ... "Bowel-associated dermatosis-arthritis syndrome after biliopancreatic diversion". ... "Bowel-associated dermatosis-arthritis syndrome in a patient with appendicitis". ... PMID 6712372 . ^ Ely PH (June 1980). "The bowel bypass syndrome: a response to bacterial peptidoglycans".
Hoyeraal–Hreidarsson syndrome Other names Progressive pancytopenia-immunodeficiency-cerebellar hypoplasia syndrome [1] This condition is inherited in an X-linked recessive manner. Specialty Medical genetics Hoyeraal–Hreidasson syndrome [2] ) is a very rare multisystem X-linked recessive disorder characterized by excessively short telomeres and is considered a severe form of dyskeratosis congenita . [2] [3] Being an X-linked disorder, Hoyeraal–Hreidasson syndrome primarily affects males. ... "Unraveling the pathogenesis of Hoyeraal–Hreidarsson syndrome, a complex telomere biology disorder" . ... "Human RTEL1 deficiency causes Hoyeraal–Hreidarsson syndrome with short telomeres and genome instability" . ... "Mutations of the RTEL1 Helicase in a Hoyeraal–Hreidarsson Syndrome Patient Highlight the Importance of the ARCH Domain".
A number sign (#) is used with this entry because of evidence that autosomal dominant dyskeratosis congenita-6 (DKCA6) and autosomal recessive dyskeratosis congenita-7 (DKCB7) are caused by heterozygous and compound heterozygous mutation, respectively, in the ACD gene (609377) on chromosome 16q22. One family with each disorder has been reported. Description Dyskeratosis congenita is a multisystem disorder caused by defective telomere maintenance. Features are variable and include bone marrow failure, nail dysplasia, oral leukoplakia, and increased risk of cancer (summary by Kocak et al., 2014). For a discussion of genetic heterogeneity of dyskeratosis congenita, see DKCA1 (127550). Clinical Features Guo et al. (2014) reported a white family in which 3 women in 3 subsequent generations had progressive bone marrow failure.
Clinical Variability Dhanraj et al. (2015) reported a 21-year-old woman with a severe form of DKCB6 and neurologic impairment consistent with a diagnosis of Hoyeraal-Hreidarsson syndrome (see 305000). She had global developmental delay apparent since infancy, profound mental retardation with absent speech, hypertonia, seizures, and hypomyelination of the central nervous system.
Description Dyskeratosis congenita is an inherited bone marrow failure syndrome classically characterized by the triad of mucosal leukoplakia, nail dysplasia, and abnormal skin pigmentation. ... Two of the DKC patients with the R252H mutation had a severe from of the disorder, with additional features including growth retardation, retinopathy, ataxia, developmental delay, and cerebellar hypoplasia, consistent with a clinical diagnosis of Hoyeraal-Hreidarsson syndrome. Nine of 57 patients with bone marrow failure and some features of DKC were found to carry mutations, including 2 with R282H, and 4 with nonsense or frameshift mutations (see, e.g., 604319.0005 and 604319.0008). ... INHERITANCE - Autosomal dominant GROWTH Height - Short stature Other - Intrauterine growth retardation - Poor growth HEAD & NECK Head - Microcephaly Ears - Deafness Eyes - Blockage of the lacrimal ducts - Epiphora - Retinopathy Mouth - Oral leukoplakia Teeth - Tooth loss RESPIRATORY Lung - Pulmonary fibrosis - Pulmonary failure GENITOURINARY Internal Genitalia (Male) - Cryptorchidism SKELETAL - Osteoporosis Limbs - Avascular necrosis of the hip (2 patients) SKIN, NAILS, & HAIR Skin - Reticular pigmentation pattern - Leukoplakia - Dry skin Nails - Dysplastic nails Hair - Premature greying - Short, fine hair - Alopecia NEUROLOGIC Central Nervous System - Speech delay - Learning difficulties - Intracranial calcifications - Cerebellar hypoplasia - Cerebellar ataxia HEMATOLOGY - Bone marrow failure - Pancytopenia - Aplastic anemia - Thrombocytopenia - Leukopenia - Increased fetal hemoglobin NEOPLASIA - Increased risk of malignancy LABORATORY ABNORMALITIES - Shortened telomeres - Decreased telomerase activity MISCELLANEOUS - Highly variable phenotype and severity, even within families - Age at onset ranges from childhood to adulthood - Phenotypic overlap with Revesz syndrome ( 268130 ) MOLECULAR BASIS - Caused by mutation in the TRF1-interacting nuclear factor 2 gene (TINF2, 604319.0001 ) ▲ Close
An X-linked syndromic intellectual disability considered to be a severe variant of dyskeratosis congenita characterized by intrauterine growth retardation, microcephaly, cerebellar hypoplasia, progressive combined immune deficiency and aplastic anemia. Epidemiology Hoyeraal-Hreidarsson syndrome (HHS) prevalence is unknown. The syndrome may be underdiagnosed due to high mortality rates. ... Differential diagnosis Differential diagnoses include dyskeratosis congenita, Revesz-Debuse syndrome, Pseudo-TORCH syndrome, Fanconi anemia and Nijmegen breakage syndrome.
Description Dyskeratosis congenita (DKC) is a bone marrow failure syndrome characterized by severely shortened telomeres and diverse clinical symptoms. ... In 1 family the heterozygous sib of a patient with autosomal recessive DKC/Hoyeraal-Hreidarsson syndrome had hypocellular bone marrow and short telomeres, but no additional features of the disorder. ... In a second family, 2 brothers had Hoyeraal-Hreidarsson syndrome associated with a heterozygous RTEL1 mutation (R1010X; 608833.0012). ... In 3 patients, including 2 sibs, with DKCB5 manifest as Hoyeraal-Hreidarsson syndrome, Le Guen et al. (2013) identified compound heterozygous mutations in the RTEL1 gene (608833.0007-608833.0010). ... In a boy with severe DKCB5 manifest as Hoyeraal-Hreidarsson syndrome, Ballew et al. (2013) identified compound heterozygous mutations in the RTEL1 gene R998X (608833.0004) and E615D (608833.0011).
Marrone et al. (2007) noted that the presence of developmental delay and cerebellar hypoplasia in the second patient was consistent with a clinical diagnosis of Hoyeraal-Hreidarsson syndrome, which is a severe variant of DKC.
Berthet et al. (1994) and Berthet et al. (1995) suggested that immunodeficiency is a feature of this syndrome. Reardon et al. (1994) suggested that this is the same condition as the autosomal recessive congenital intrauterine infection-like syndrome, or pseudo-TORCH syndrome (251290). Aalfs and Hennekam (1995) described several differences between the 2 syndromes. Patients with Hoyeraal-Hreidarsson syndrome show only growth retardation and microcephaly in the first months of life, whereas those with the pseudo-TORCH syndrome have symptoms resembling TORCH infection shortly after birth, including hepatosplenomegaly. Furthermore, in the intrauterine infection-like syndrome, the neonatally present thrombocytopenia resolves within a year if the child survives, whereas in the Hoyeraal-Hreidarsson syndrome the first symptoms of pancytopenia do not occur before the age of 5 months and continue to increase for years. The cerebellum is proportionately small in Hoyeraal-Hreidarsson syndrome, whereas the cerebral abnormalities are more severe in the pseudo-TORCH syndrome. ... This child had striking features of both Hoyeraal-Hreidarsson syndrome and X-linked dyskeratosis congenita.
Nicolau–Balus syndrome Specialty Dermatology Nicolau–Balus syndrome is a cutaneous condition characterized by syringomas and milia . [1] See also [ edit ] Parry–Romberg syndrome List of cutaneous conditions List of cutaneous neoplasms associated with systemic syndromes References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).
WPW syndrome is a heart condition present at birth (congenital heart defect). ... Rarely, WPW syndrome may lead to sudden cardiac death in children and young adults. ... WPW syndrome may also be called preexcitation syndrome. ... The most common arrhythmia seen with WPW syndrome is supraventricular tachycardia. ... Rarely, WPW syndrome is passed down through families (inherited).
She reported that closed bite and dental open are two of the syndrome's variants. [A] [7] The treatment for young patients troubled by long face syndrome is to halt and control descent of the lower jaw and to prevent the eruption of posterior teeth . In severe cases of deformity, a mixture of orthodontics and orthognathic surgery may be the only effective solution. [8] [9] The long-term (more than 6 years) effectiveness of surgical treatments for long face syndrome has been subject to study. [10] "In the American literature, the terms long-face syndrome and short-face syndrome are often used." ... However, in the opinion of Hugo Obwegeser , there is no medical justification for naming them as a " syndrome " — the signs and symptoms do not meet the definitional threshold. [11] : 22 There is controversy concerning the use of the descriptor "long-face syndrome." ... "Orthodontic diagnosis of long face syndrome" . General Dentistry . 44 (4): 348–351. ... (September 1990). "The long face syndrome and impairment of the nasopharyngeal airway" .
Symptom severity can vary from person to person and depends on the specific type of Ehlers-Danlos syndrome that you have. The most common type is called hypermobile Ehlers-Danlos syndrome. Vascular Ehlers-Danlos syndrome People who have vascular Ehlers-Danlos syndrome often share distinctive facial features of a thin nose, thin upper lip, small earlobes and prominent eyes. ... For hypermobile Ehlers-Danlos syndrome, the most common form, there is no genetic testing available. ... Lifestyle and home remedies If you have Ehlers-Danlos syndrome, it's important to prevent injuries. ... Knowing more about Ehlers-Danlos syndrome can help you take control of your condition.
Giunta et al [2005a] convincingly demonstrated that Nevo syndrome is part of the spectrum of EDS VI; thus, the term "Nevo syndrome" does not refer to a distinct disorder but is now incorporated into kEDS. ... Differential Diagnosis Other forms of Ehlers-Danlos syndrome. Kyphoscoliotic Ehlers-Danlos syndrome (kEDS) has some overlapping clinical features with other forms of EDS, particularly classic EDS and vascular EDS. ... This condition, described in 26 individuals to date, is classified as kEDS- FKBP14 in the 2017 International Classification of the Ehlers-Danlos Syndromes [Malfait et al 2017]. See FKBP14 -Related Kyphoscoliotic EDS. ... Corneal disorder. Brittle cornea syndrome (BCS) is associated with corneal rupture following minor trauma (OMIM 229200 and 614170). ... Disorders with early-onset hypotonia. Many syndromic and metabolic disorders include early-onset hypotonia.
Summary Clinical characteristics. Classic Ehlers-Danlos syndrome (cEDS) is a connective tissue disorder characterized by skin hyperextensibility, atrophic scarring, and generalized joint hypermobility (GJH). ... Diagnosis Suggestive Findings The diagnosis of classic Ehlers-Danlos syndrome (cEDS) can be suspected based on clinical examination and family history. ... Molecular Genetic Testing Used in Classic Ehlers-Danlos Syndrome (cEDS) View in own window Gene 1, 2 Proportion of cEDS Attributed to Pathogenic Variants in Gene Proportion of Pathogenic Variants 3 Detectable by Method Sequence analysis 4 Gene-targeted deletion/duplication analysis 5 COL1A1 <1% 6 100% 6 None reported 6 COL5A1 75%-78% 7 99% 7 1% 7 COL5A2 14% 7 100% 7 Unknown 8 Unknown <10% 7 NA 1. ... Clinical Characteristics Clinical Description Classic Ehlers-Danlos syndrome (cEDS) is a connective tissue disorder characterized by skin hyperextensibility, abnormal wound healing, and generalized joint hypermobility. ... Differential Diagnosis Other forms of Ehlers-Danlos syndrome (EDS) should be considered in individuals with easy bruising, joint hypermobility, and/or chronic joint dislocation.
"Dystonia in the joint hypermobility syndrome (a.k.a. Ehlers- Danlos syndrome, hypermobility type)" . ... PMID 23154631 . ^ "Ehlers-Danlos syndrome" . Genetic Home Reference . Retrieved 4 April 2018 . ^ a b c d e "Ehlers–Danlos Syndrome" . ... "Heart rate, conduction and ultrasound abnormalities in adults with joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type" . ... PMID 24752348 . ^ Wenstrup RJ, et al. (2001). The Ehlers–Danlos Syndromes: Management of Genetic Syndromes . pp. 131–149. ^ Grigoriou E, Boris JR, Dormans JP (February 2015). "Postural orthostatic tachycardia syndrome (POTS): association with Ehlers-Danlos syndrome and orthopaedic considerations" .
Ehlers-Danlos syndromes (EDS) are a group of inherited connective tissue disorders caused by abnormalities in the structure, production, and/or processing of collagen .
Unsourced material may be challenged and removed. Find sources: "Irlen syndrome" – news · newspapers · books · scholar · JSTOR ( August 2011 ) ( Learn how and when to remove this template message ) ( Learn how and when to remove this template message ) Irlen syndrome Pseudomedical diagnosis Risks Nocebo Irlen syndrome , occasionally referred to as scotopic sensitivity syndrome ( SSS ) or Meares-Irlen syndrome , [1] is a proposed disorder of vision or image-processing in the brain. ... Irlen syndrome, for example, seems to include a broader array of symptoms, including severe variants of the core condition. ... The second, which focused primarily on Irlen syndrome, found compelling evidence of unique brain function linked to the syndrome. ... When patients exhibiting Meares Irlen Syndrome were treated with vision therapy, their symptoms were relieved. ... S2CID 15631430 . ^ https://sciencebasedmedicine.org/irlen-syndrome/ ^ Ritchie, S. J.; Della Sala, S.; McIntosh, R.