Proper counselling and surgery are required to enable the normal adult life of these individuals. [5] [6] Contents 1 Classification 2 Signs and symptoms 3 Associated syndromes 3.1 Ectodermal dysplasia 3.2 Poland's syndrome 3.3 Al Awadi/Raas-Rothschild syndrome 3.4 Scalp-ear-nipple syndrome 4 Mechanism 5 Causes 6 Genetics 7 Management 7.1 Breast reconstruction 7.2 Nipple areola relocation 8 See also 9 References 10 External links Classification [ edit ] Amastia can be either iatrogenic or congenital. [1] The congenital amastia are further divided into syndromic type and non-syndromic type respectively. As the definition suggests, syndromic amastia is often associated with obvious symptoms. ... Associated syndromes [ edit ] Amastia, particularly if it is bilateral, often related to various syndromes, including ectodermal dysplasia and Poland's syndrome, which is characterised by anomalies of underlying mesoderm and abnormal pectoral muscle respectively. [8] Other syndromes, such as FIG4 associated Yunis Varon syndrome (MIM 216340), acro-der-mato-ungual-lacrimal-tooth (ADULT) syndrome, TP63 associated limb mammary syndrome (MIM 603543), TBX3 associated ulnar syndrome (MIM 181450) and KCTD1 associated scalp-ear-nipple syndrome (MIM 181270) have also been clinically observed. [3] Ectodermal dysplasia [ edit ] Ectodermal dysplasia is commonly associated with syndromic amastia. ... This suggests people with amastia should have a comprehensive skin test to exclude this syndrome. [8] Poland's syndrome [ edit ] Poland's syndrome is a genetic disorder associated with abnormal breast development. The prevalence rate of this syndrome is approximately 1 in 20000 to 30000.
Absence of the mammary gland Not to be confused with Amaziah . Amazia refers to a condition where one or both of the mammary glands is absent (the nipple and areola remain present). [1] This may occur either congenitally or iatrogenically (typically the result of surgical removal and/or radiation therapy). Amazia can be treated with breast implants . Amazia differs from amastia (the complete absence of breast tissue, nipple, and areola), although the two conditions are often (erroneously) thought to be identical. The terms "amazia" and "amastia" are thus often used interchangeably, even though the two conditions are medically different. See also [ edit ] Amastia Athelia Micromastia References [ edit ] ^ Ozsoy Z, Gozu A, Ozyigit MT, Genc B (2007). "Amazia with midface anomaly: case report". Aesthetic Plast Surg . 31 (4): 392–4. doi : 10.1007/s00266-006-0251-0 .
Bilateral absence of the breasts may occur as an isolated anomaly or may be associated with a syndrome or a cluster of other anomalies, including anhidrotic ectodermal dysplasia (305100) or Poland syndrome (173800) (summary by Papadimitriou et al., 2009).
Bilateral absence of the breasts may occur as an isolated anomaly or may be associated with a syndrome or a cluster of other anomalies, including anhidrotic ectodermal dysplasia (305100) and Poland syndrome (173800) (summary by Papadimitriou et al., 2009).
Epidermolytic hyperkeratosis is a skin disorder that is present at birth. Affected babies may have very red skin (erythroderma) and severe blisters. Because newborns with this disorder are missing the protection provided by normal skin, they are at risk of becoming dehydrated and developing infections in the skin or throughout the body (sepsis). As affected individuals get older, blistering is less frequent, erythroderma becomes less evident, and the skin becomes thick (hyperkeratotic ), especially over joints, on areas of skin that come into contact with each other, or on the scalp or neck. This thickened skin is usually darker than normal. Bacteria can grow in the thick skin, often causing a distinct odor.
Transient Headache and Neurologic Deficits With Cerebrospinal Fluid Lymphocytosis (HaNDL syndrome) is a headache disorder in which individuals experience severe to moderate headache attacks, neurological symptoms, and an increase in the amount of a type of white blood cell in the cerebrospinal fluid (lymphocytic pleocytosis). HaNDL syndrome is most often diagnosed in adulthood, although individuals with ages ranging from 7 to 52 have been reported with this condition. The most common neurological symptoms associated with HanDL syndrome include: weakness on one side of the body (hemiparesis), a feeling of altered sensation down one side of the body (hemisensory disturbances), and a loss of the ability to understand or express speech (aphasia). These symptoms usually last between 15 minutes and 2 hours. The cause of HaNDL syndrome is not well understood; however, researchers have suggested a few potential causes, including migraines, inflammation of the tissue that lines the brain and spinal cord, and viral infections. For some individuals, HaNDL syndrome may resolve without treatment, while for others, management of headache and neurological symptoms may be needed.
Swyer syndrome is a genetic condition affecting sexual organ development, classified as a disorder of sex development (DSD). In Swyer syndrome, people with one X chromosome and one Y chromosome, normally present in males, are born with female external genitalia and underdeveloped gonads (ovaries or testes) known as streak gonads. Most people with Swyer syndrome are raised as females. Adolescents with this condition do not go through normal puberty and are infertile. Some cases of Swyer syndrome are caused by genetic alterations (mutations) in one of several genes, but in some cases the cause is unknown. ... Treatment includes removal of streak gonads to prevent cancer and hormone replacement therapy from puberty onward. While women with Swyer syndrome are infertile, they may become pregnant with the use of donated eggs.
A number sign (#) is used with this entry because this form of 46,XY sex reversal is caused by heterozygous mutation in the NR5A1 gene (184757) on chromosome 9q33. For a discussion of genetic heterogeneity of 46,XY sex reversal, see SRXY1 (400044). Nomenclature As a result of discussions at the International Consensus Conference on Intersex, Lee et al. (2006) proposed the term 'disorder(s) of sex development' (DSD) to replace the previously used terms 'pseudohermaphroditism,' 'intersex,' and 'sex reversal.' Clinical Features Achermann et al. (1999) described a phenotypically female patient who presented with primary adrenal failure in the first 2 weeks of life. Her karyotype was XY, and a presumptive diagnosis of congenital lipoid adrenal hyperplasia (201710) was made.
They are chromatin-negative and are usually of normal stature, without the somatic stigmata of Turner syndrome (see 163950) (summary by Mann et al., 1983). ... The 2 sisters had bilateral streak ovaries that were grossly and histologically similar to those seen in 45,X gonadal dysgenesis (Turner syndrome); their affected cousin had gonads resembling the streaks grossly, but histopathology revealed bilateral gonadoblastoma.
A number sign (#) is used with this entry because of evidence that this form of 46,XY sex reversal is caused by heterozygous mutation in the MAP3K1 gene (600982) on chromosome 5q11. For a discussion of genetic heterogeneity of 46,XY sex reversal, see SRXY1 (400044). Clinical Features Sternberg et al. (1968) reported 3 cases of 46,XY females, each in a different sibship of a family connected through normal females (proposita, maternal cousin, and maternal aunt). Espiner et al. (1970) studied a New Zealand kindred of European descent in which 5 phenotypic females from 3 sibships had a normal XY karyotype in all stem lines examined and pure gonadal dysgenesis, with bilateral streak gonads verified at laparotomy. The proband presented at 21 years of age because she was still growing, and had grown 1.9 cm in height over the past 6 months.
A number sign (#) is used with this entry because of evidence that 46,XY sex reversal-5 (SRXY5) is caused by compound heterozygous mutation in the CBX2 gene (602770) on chromosome 17q25. One such patient has been reported. For a discussion of genetic heterogeneity of 46,XY sex reversal, see SRXY1 (400044). Clinical Features Biason-Lauber et al. (2009) reported a child who, despite prenatally determined 46,XY karyotype, presented at birth as a phenotypically normal female. The karyotype was confirmed antenatally. She had low testosterone at 2 years of age that failed to increase after stimulation with chorionic gonadotropin (CG; 118860). Laparoscopy performed at 4.5 years to evaluate and potentially remove the gonads showed 2 ovaries of normal appearance.
Nomenclature The designation 'Swyer syndrome' refers to 46,XY complete gonadal dysgenesis. ... Rushton (1979) pointed out that the streak gonads of this disorder differ from those of the 45,X Turner syndrome in the presence of calcification and the increased hazard of gonadoblastoma. ... The 2 patients reported by Disteche et al. (1986) had some signs of Turner syndrome, including congenital lymphedema and primary amenorrhea with streak gonads, but were of normal height. ... Moreira-Filho et al. (1979) suggested that there are 3 forms of Swyer syndrome (defined as streak gonads without other somatic features of the Turner syndrome and with a normal 46,XY karyotype). (1) Sporadic testicular agenesis syndrome (STAS) corresponds to H-Y negative Swyer syndrome. (2) Familial testicular agenesis syndrome (FTAS) is H-Y negative Swyer syndrome showing an X-linked recessive pedigree pattern. ... Passarge and Wolf (1981) pointed out that there are 2 groups of patients with XY gonadal dysgenesis (Swyer syndrome) and that each of these may be heterogeneous.
A number sign (#) is used with this entry because of evidence that dosage-sensitive sex reversal is due to duplication of the DAX1 gene (NR0B1; 300473) on chromosome Xp21.3-p21.2. For a discussion of genetic heterogeneity of 46,XY sex reversal, see SRXY1 (400044). Clinical Features The existence of an X-specific gene involved in human sex determination was first postulated by German et al. (1978), on the basis of a family with an apparent X-linked mode of inheritance of 46,XY gonadal dysgenesis. A number of families with X-linked recessive (or sex-limited autosomal dominant) transmission of the disorder were reported thereafter (reviewed by Fechner et al., 1993). Bardoni et al. (1994) studied 4 patients with 46,XY sex reversal, who were raised as girls due to their having either ambiguous or female external genitalia.
An alternative designation they used for this condition was familial testicular agenesis syndrome (FTAS). They pointed to the cases of Haseltine and Ohno (1981) and Ghosh et al. (1978) as likewise representing S mutations. Cases of H-Y antigen-positive XY gonadal dysgenesis differ from the cases just mentioned by the findings in the gonads which show testicular primordia; hence the term familial testicular dysgenesis syndrome (FTDS) suggested by Moreira-Filho et al. (1979).
A number sign (#) is used with this entry because 9p24.3 deletions have been observed in patients with 46,XY sex reversal without other features of the chromosome 9p deletion syndrome (158170). For a discussion of genetic heterogeneity of 46,XY sex reversal, see SRXY1 (400044). ... Veitia et al. (1998) reported studies of 2 patients with 46,XY complete or partial gonadal dysgenesis associated with 9p deletions but without the rest of the features of monosomy 9p syndrome. Mapping Shan et al. (2000) reviewed the accumulating evidence that haploinsufficiency of a dosage-sensitive gene(s) on 9p24.3 is responsible for the failure of testicular development and feminization in XY patients with monosomy for 9p. ... Barbaro et al. (2009) noted that separate regions of deletion on chromosome 9 had been identified for 46,XY gonadal dysgenesis and monosomy 9p deletion syndrome: 9p24.3, extending from the DMRT genes to the telomere for the former, and 9p23-p22.3 for the latter.
Stature is normal or above normal, and features of Turner syndrome (see this term) are absent. Etiology Although the etiology is not completely understood, 46,XY CGD results from failure of testicular development due to disruption of the underlying genetic pathways and several genes have been implicated: SRY (gene deletion of or loss-of-function mutations; Yp11.3), NR5A1 (9q33) and DHH (homozygous or compound heterozygous mutations; 12q13.1). ... Differential diagnosis The differential diagnosis should include hypergonadotropic ovarian dysgenesis (46,XX GD) and all forms of syndromic 46,XY CGD (for example, Frasier syndrome, campomelic dysplasia and 46,XY DSD with adrenal insufficiency; see these terms). Antenatal diagnosis Prenatal diagnosis is feasible for families in which the genetic anomaly has been confirmed but is only recommended in syndromic cases. Genetic counseling Although some cases of 46,XY CGD occur sporadically, genetic counseling may be offered to affected families and should be adapted depending on the mode of inheritance associated with the genetic anomaly identified.
Nuclear gene-encoded Leigh syndrome is a progressive neurological disease . ... Other signs and symptoms may include an increase in the heart muscle size ( hypertrophic cardiomyopathy ); excessive body hair (hypertrichosis); anemia; kidney or liver problems; and lung or heart failure. Nuclear gene-encoded Leigh syndrome (and Leigh-like syndrome, a term used for cases with similar features but that do not fulfill the diagnostic criteria for Leigh syndrome) may be caused by mutations in any of several genes and can be inherited in an autosomal recessive or X-linked manner. While treatment for some cases of Leigh-like syndrome may be available, management is generally supportive and focuses on the symptoms present.
2q23.1 microdeletion syndrome is a rare chromosome disorder. ... Children affected by 2q23.1 microdeletion syndrome may also have low muscle tone (hypotonia), slow weight gain, and may be shorter than family members. 2q23.1 deletion syndrome is caused by the loss of a small piece of DNA in one copy of chromosome 2, one of the 23 pairs of chromosomes in each cell in our bodies. Most cases of 2q23.1 deletion syndrome are de novo , which means the deletion was not passed down from either parent. Diagnosis of 2q23.1 microdeletion syndrome may be suspected by symptoms but is confirmed by genetic testing.
Behavior problems (observed in MBD5 haploinsufficiency and other forms of syndromic autism) [Talkowski et al 2011, Hodge et al 2014] include the following: Short attention span and autistic-like behaviors (gaze avoidance, inattention, and repetitive behaviors) [Tan et al 2014] and stereotypic and repetitive behaviors (e.g., teeth grinding, chewing of the hands, and repetitive hand movements) are seen in more than 80% of affected individuals. ... This disorder was initially called 2q23.1 deletion syndrome; however, delineation of the smallest region of overlap among individuals with a 2q23.1 deletion confirmed that haploinsufficiency of MBD5 is responsible for the disorder. ... Significant similarity in phenotypic features is observed between MBD5 haploinsufficiency and other neurodevelopmental syndromes. Specific disorders to consider in the differential diagnosis of MBD5 haploinsufficiency include the following: Smith-Magenis syndrome (SMS) Angelman syndrome (AS) Pitt-Hopkins syndrome Rett syndrome Fragile X syndrome Kleefstra syndrome Koolen-De Vries syndrome Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with MBD5 haploinsufficiency, the following evaluations are recommended: Physical and neurologic examination Developmental assessment including assessment by a physical therapist and/or occupational therapist Speech evaluation including feeding evaluation and nutrition consultation as needed Hearing assessment (as part of the routine evaluation of children with speech delay) EEG if seizures are suspected Cardiac evaluation Sleep history Behavioral assessment Consultation with a clinical geneticist and/or genetic counselor Treatment of Manifestations A multidisciplinary approach to manage the features and specific issues identified is recommended.
Eagle syndrome is characterized by recurrent pain in the middle part of the throat (oropharynx) and face. "Classic Eagle syndrome" is typically seen in patients after throat trauma or tonsillectomy. ... Other symptoms may include difficulty swallowing, feeling of something stuck in the throat, tinnitus , and neck or facial pain. A second form of Eagle syndrome unrelated to tonsillectomy causes compression of the vessel that carries blood to the brain, neck, and face (carotid artery). This form can cause headache and dizziness. Eagle syndrome is due to a calcified stylohyoid ligament or an elongated styloid process. ... The mainstay treatment for Eagle syndrome is surgery to shorten the styloid process (styloidectomy).
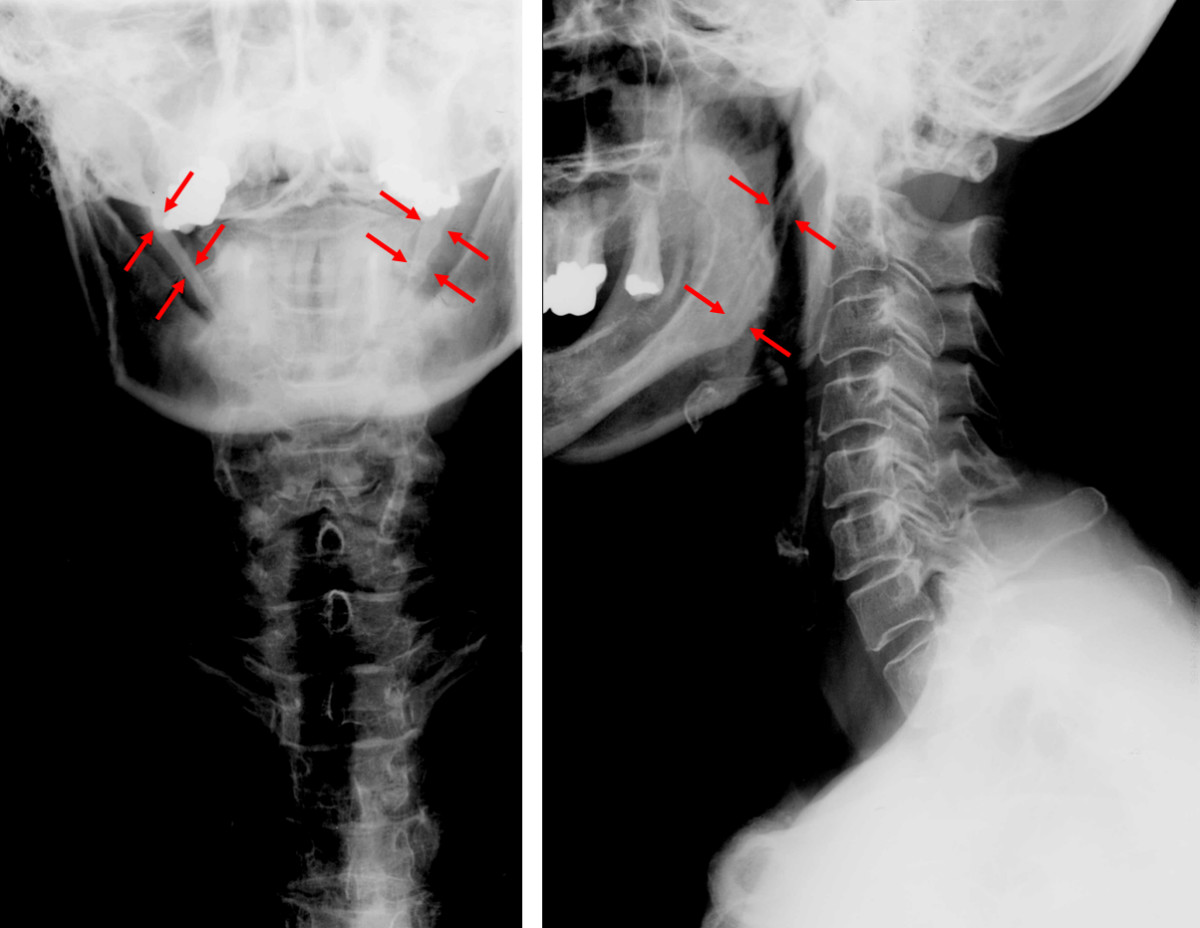
Eagle syndrome Other names Styloid syndrome Anteroposterior and lateral radiographs of cervical spine showing ossification of the stylohyoid ligament on both sides Eagle syndrome (also termed stylohyoid syndrome , [1] styloid syndrome , [2] styloid-stylohyoid syndrome , [2] or styloid–carotid artery syndrome ) [3] is a rare condition commonly characterized but not limited to sudden, sharp nerve-like pain in the jaw bone and joint , back of the throat , and base of the tongue, triggered by swallowing, moving the jaw, or turning the neck. ... "Styloidogenic jugular venous compression syndrome: diagnosis and treatment: case report". ... PMID 19881063 . ^ Karam C, Koussa S (December 2007). "[Eagle syndrome: the role of CT scan with 3D reconstructions]" . ... "[Elongated styloid process (Eagle's syndrome): literature review and a case report]". Agri (in Turkish). 17 (2): 23–5. PMID 15977090 . ^ "Eagle syndrome" . rarediseases . Genetic and rare diseases information center .
DICER1-related pleuropulmonary blastoma cancer predisposition syndrome causes a moderately increased risk for certain cancers and tumors. ... Cysts in the kidneys (cystic nephroma) are also associated with DICER1 syndrome. These cysts typically develop in childhood, but do not usually cause any health problems. Women with DICER1 syndrome are at an increased risk for Sertoli-Leydig tumors of the ovaries. DICER1 syndrome is also associated with goiter (multiple fluid-filled or solid tumors in the thyroid gland). These goiters typically occur in adulthood and most often do not cause symptoms. This syndrome is caused by mutations in the DICER1 gene.
Description Pleuropulmonary blastoma (PPB) is a rare pediatric tumor of the lung that arises during fetal lung development and is often part of an inherited cancer syndrome (Hill et al., 2009). PPBs contain both epithelial and mesenchymal cells. ... In approximately 35% of families in which a child has PPB, the patient or a family member manifests 1 or more additional conditions from an unusual array of dysontogenetic-dysplastic and malignant conditions, known as the 'PPB family tumor and dysplasia syndrome' (PPBFTDS). Cystic nephroma, which are benign lesions of the kidney, are found in 9 to 10% of family members affected by PPB (summary by Bahubeshi et al., 2010). Larger studies have shown that DICER1 mutations are associated with a variety of tumor types, indicating that this disorder can be considered a tumor predisposition syndrome (summary by Slade et al., 2011). ... Slade et al. (2011) proposed the designation 'DICER1 syndrome.' Analysis of tumor tissue and cancer cell lines indicated that somatic DICER1 mutations are unlikely to contribute to tumor pathogenesis.
The tumor is often part of pleuropulmonary blastoma family tumor and dysplasia syndrome. It can also be associated with multilocular cystic nephroma or other neoplasms.
Glucose transporter type 1 deficiency syndrome (GLUT1 deficiency syndrome) is an inherited condition that affects the nervous system. ... Approximately 10% of affected people have the "non-epileptic" form of GLUT1 deficiency syndrome which is associated with all the typical symptoms of the condition without seizures. GLUT1 deficiency syndrome is caused by changes (mutations) in the SLC2A1 gene and is inherited in an autosomal dominant manner. Although there is currently no cure for GLUT1 deficiency syndrome, a special diet (called a ketogenic diet ) may help alleviate symptoms.
The phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS) is now known to be a continuum that includes the classic phenotype as well as paroxysmal exercise-induced dyskinesia and epilepsy (previously known as dystonia 18 [DYT18]) and paroxysmal choreoathetosis with spasticity (previously known as dystonia 9 [DYT9]), atypical childhood absence epilepsy, myoclonic astatic epilepsy, and paroxysmal non-epileptic findings including intermittent ataxia, choreoathetosis, dystonia, and alternating hemiplegia. ... Diagnosis Suggestive Findings Glucose transporter type 1 deficiency syndrome (Glut1 DS) should be suspected in individuals with one of the following two phenotypes. ... Clinical Characteristics Clinical Description Glucose transporter type 1 deficiency syndrome (Glut1 DS) usually presents as either classic Glut1 DS (~90% of affected individuals) or, more rarely, non-classic Glut1 DS (~10% of affected individuals), which comprises a broad phenotypic spectrum. ... Differential Diagnosis The differential diagnosis of glucose transporter type 1 deficiency syndrome (Glut1 DS) includes the following: Other causes of neuroglycopenia including conditions causing chronic or intermittent hypoglycemia (e.g., familial hyperinsulinism) All causes of neonatal seizures and of acquired microcephaly; in particular, early presentations of Rett syndrome, Angelman syndrome, and infantile forms of neuronal ceroid-lipofuscinosis Opsoclonus-myoclonus syndrome [Pike 2013, Pearson et al 2017] Cryptogenic epileptic encephalopathies with developmental delays Familial epilepsies with autosomal dominant transmission Episodes of paroxysmal neurologic dysfunction responsive to or preventable by carbohydrate intake, especially when manifesting as alternating hemiparesis, ataxia, cognitive dysfunction, or seizures Movement disorders including dystonia (see Dystonia Overview) Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs of an individual diagnosed with glucose transporter type 1 deficiency syndrome (Glut1 DS), the following evaluations are recommended if they have not already been completed: EEG (pre-prandial and post-prandial tracings) Brain imaging, including FDG-PET in selected individuals Neuropsychological assessment Consultation with a clinical geneticist and/or genetic counselor Treatment of Manifestations Ketogenic diet.
Allelic disorders with overlapping features include GLUT1 deficiency syndrome with pseudohyperkalemia and hemolysis (608885), GLUT1 deficiency syndrome-2 (GLUT1DS2; 612126), dystonia-9 (DYT9; 601042), and idiopathic generalized epilepsy-12 (EIG12; 614847). Description GLUT1 deficiency syndrome-1 is a neurologic disorder showing wide phenotypic variability. ... Rotstein et al. (2010) provided further details of a patient with autosomal recessive GLUT1 deficiency syndrome reported by Wang et al. (2000). ... In the family reported by Brockmann et al. (2001), the GLUT1 deficiency syndrome affected 5 members over 3 generations. The syndrome behaved as an autosomal dominant, with 1 instance of father-to-son transmission.
Glucose transporter type 1 (GLUT1) deficiency syndrome is characterized by an encephalopathy marked by childhood epilepsy that is refractory to treatment, deceleration of cranial growth leading to microcephaly, psychomotor retardation, spasticity, ataxia, dysarthria and other paroxysmal neurological phenomena often occurring before meals. ... Genetic counseling GLUT1 deficiency syndrome is transmitted as an autosomal dominant trait and in these cases the affected parent presents with a mild form of the disease.
Retrieved 2018-01-31 . ^ a b "GLUT1 deficiency syndrome" . Genetics Home Reference . Retrieved 10 October 2011 . ^ a b c d e f g h i "Brochures" . ... "Glucose Transporter Type 1 Deficiency Syndrome". GeneReviews . CS1 maint: multiple names: authors list ( link ) ^ a b c Reference, Genetics Home. "GLUT1 deficiency syndrome" . Genetics Home Reference . Retrieved 2018-01-25 . ^ a b c d e "Reaching for a brighter future" (PDF) . ... "Paroxysmal eye–head movements in GLUT1 deficiency syndrome" . Neurology . 88 (17): 1666–1673. doi : 10.1212/WNL.0000000000003867 . ... ISSN 1090-3798 . ^ Reference, Genetics Home. "GLUT1 deficiency syndrome" . Genetics Home Reference . Retrieved 2017-06-15 . ^ a b "Professional Guide" (PDF) .
Kasabach–Merritt syndrome Other names Hemangioma-thrombocytopenia syndrome Specialty Hematology Kasabach–Merritt syndrome , also known as hemangioma with thrombocytopenia [1] is a rare disease , usually of infants , in which a vascular tumor leads to decreased platelet counts and sometimes other bleeding problems , [2] which can be life-threatening. [3] It is also known as hemangioma thrombocytopenia syndrome . ... ISBN 978-0-7216-2921-6 . ^ a b c d e Hall G (2001). "Kasabach–Merritt syndrome: pathogenesis and management". ... "Infants with Kasabach–Merritt syndrome do not have "true" hemangiomas". ... PMID 3278084 . ^ a b c d Kasabach-Merritt Syndrome at eMedicine ^ Larsen, EC; Zinkham, WH; Eggleston, JC; Zitelli, BJ (June 1987). "Kasabach-Merritt syndrome: therapeutic considerations". Pediatrics . 79 (6): 971–80.
Animal Model In a mouse model of Kasabach-Merritt syndrome, Verheul et al. (1999) stimulated platelet production using Peg-rHuMGDF and observed a 7- to 8-fold increase in platelet counts and a significantly increased survival, with 50% of treated animals alive at 1 month versus none of the untreated controls.
Hemangioma thrombocytopenia syndrome is characterized by profound thrombocytopenia in association with two rare vascular tumors: kaposiform hemangioendotheliomas and tufted angiomas .
Kasabach-Merritt syndrome (KMS), also known as hemangioma-thrombocytopenia syndrome, is a rare disorder characterized by profound thrombocytopenia, microangiopathic hemolytic anemia, and subsequent consumptive coagulopathy in association with vascular tumors, particularly kaposiform hemangioendothelioma or tufted angioma.
Anton–Babinski syndrome is the right-hemisphere equivalent of Gerstmann syndrome and it is due to non-dominant inferior parietal lobule damage. ... Causes [ edit ] Why patients with Anton syndrome deny their blindness is unknown, although there are many theories. ... The syndrome features prominently in the Rupert Thomson novel The Insult . ... "Anton syndrome as a result of MS exacerbation" . ... R.; Syed, E. H. (2007). "Anton-Babinski Syndrome in a Child with Early-stage Adrenoleukodystrophy".
White matter hypoplasia-corpus callosum agenesis-intellectual disability syndrome is a very rare neurological condition. ... It can be either asymptomatic or associated with intellectual disability, epilepsy, or psychiatric syndromes. It can be part of several genetic syndromes, such as Aicardi syndrome , Andermann syndrome and Apert syndrome , trisomies 13 , 18 ; or result from metabolic causes; drugs (cocaine); or viral infection (influenza).
A rare, genetic, multiple congenital anomalies/dysmorphic syndrome characterized by severe white matter hypoplasia, corpus callosum agenesis or extreme hypoplasia, severe intellectual disability, failure to thrive and minor midline facial dysmorphism (including hypertelorism, broad nasal root, micrognathia).
Shapiro syndrome Other names Spontaneous periodic hypothermia Specialty Neurology Shapiro syndrome is an extremely rare disorder consisting of paroxysmal hypothermia (due to hypothalamic dysfunction of thermoregulation ), hyperhydrosis (sweating), and agenesis of the corpus callosum with onset typically on adulthood. ... Very little is known about the disease due to the small number of people affected. [1] References [ edit ] ^ Shapiro Syndrome , Genetic and Rare Diseases Information Center (GARD), National Institutes of Health Further reading [ edit ] "Shapiro syndrome" Shenoy C. QJM . 2008 Jan;101(1):61-2. PMID 18203725 "Shapiro syndrome with hypothalamic hypothyroidism" Arkader R, Takeuchi CA. ... PMID 18641886 "Subtotal corpus callosum agenesis with recurrent hyperhidrosis-hypothermia (Shapiro syndrome)" Tambasco N, Corea F, Bocola V.
Etiology The exact pathophysiological mechanism for this syndrome is still not understood. Postulated mechanisms include hypothalamic dysfunction, neurochemical abnormalities, inflammatory processes, and epileptic activity.
Shapiro syndrome is a rare neurological disease characterized by recurrent episodes of excessive sweating and hypothermia along with agenesis of the corpus callosum .
Meacham syndrome is a multiple malformation syndrome characterized by congenital diaphragmatic abnormalities, genital defects and cardiac malformations. ... Etiology Mutations in the WT1 gene have been identified in some patients with Meacham syndrome. Mutations in the same gene have previously been detected in patients with Denys-Drash syndrome (see this term). ... Differential diagnosis The main differential diagnosis comprises Denys-Drash syndrome, an allelic disorder with overlapping clinical features. Beckwith-Wiedemann, Simpson-Golabi-Behmel and Perlman syndromes should also be considered (see these terms).
A number sign (#) is used with this entry because some patients with Meacham syndrome have mutations in the WT1 gene (607102). See also Denys-Drash syndrome (194080), an allelic disorder with overlapping clinical features. ... The authors stated that this was the fifth reported case of Meacham syndrome. Suri et al. (2007) reported detailed clinical features of 8 patients, including 2 half-sibs, with Meacham syndrome. ... None had renal mesangial sclerosis or Wilms tumor, thus excluding a diagnosis of Denys-Drash syndrome. Molecular Genetics In 2 patients with Meacham syndrome, Suri et al. (2007) identified heterozygous mutations in the WT1 gene (R366W, 607102.0026 and R394W, 607102.0003); neither patient had congenital heart defects. ... Suri et al. (2007) noted that both WT1 mutations had been identified in patients with Denys-Drash syndrome, illustrating the phenotypic overlap between the 2 disorders.
For Harry Benjamin syndrome, see Transsexual § Terminological variance . Benjamin syndrome Other names Benjamin anemia Benjamin Syndrome is a type of multiple congenital anomaly / intellectual disability (MCA/MR) syndrome. ... Verh Deut Ges Kinderh , 1911,119-24. ^ Jablonski, Stanley (1991). Jablonski's dictionary of syndromes & eponymic diseases. Krieger Pub. ... ISBN 978-0-89464-224-1 External links [ edit ] Benjamin syndrome via National Library of Medicine.
Binder's syndrome Specialty Medical genetics Binder's Syndrome / Binder Syndrome (Maxillo-Nasal Dysplasia) is a developmental disorder primarily affecting the anterior part of the maxilla and nasal complex (nose and jaw). It is a rare disorder and the causes are unclear. The characteristics of the syndrome are typically visible. The syndrome involves hypoplasia of variable severity of cartilaginous nasal septum and premaxilla. ... "Maxillo-Nasal Dysplasia (Binder Syndrome): Antenatal Discovery and Implications"], Fetal Diagnosis and Therapy , 2005.
Binder syndrome is a rare developmental anomaly, affecting primarily the anterior part of the maxilla and nasal complex. Epidemiology Binder syndrome occurs in less than one birth in 10,000 but is probably underdiagnosed. ... Etiology The etiology and pathogenesis of Binder syndrome remains uncertain. Differential diagnosis Phenocopies of Binder syndrome have been described in children exposed in utero to phenytoin or to vitamin K deficiency, being induced either by drug (anticoagulants) or by biliary lithiasis. Some authors consider Binder syndrome as an allelic form of chondrodysplasia punctata. Others suggest that Binder type maxillonasal dysplasia does not represent a distinct disease entity or syndrome, but rather is a nonspecific abnormality of the nasomaxillary regions.
Critical features of the syndrome appear to be midfacial hypoplasia, lack of anterior nasal spine, and malocclusion. Sheffield et al. (1989) reviewed 103 cases of chondrodysplasia punctata (CDP; see 215100) seen in Melbourne over a 20-year period. They concluded that Binder syndrome should be classified as a mild form of CDP. ... Olow-Nordenram and Radberg (1984) reported the syndrome in mother and daughter also. Roy-Doray et al. (1997) described Binder syndrome in a mother and her son. ... Photographs of both patients as infants and of the mother at age 19 were provided. The Keutel syndrome (245150) shares some features with Binder syndrome, such as midfacial hypoplasia. Munroe et al. (1999) suggested that Binder syndrome might be an allelic variant of Keutel syndrome, which has been shown to be due to mutations in the gene encoding matrix Gla protein (MGP; 154870).
Zechi-Ceide syndrome Other names Occipital atretic cephalocele-unusual facies-large feet syndrome Symptoms facial abnormalities, large feet and mental deficiency Zeichi-Ceide syndrome is a rare disease discovered in 2007. ... The investigators suspected the symptoms were caused by autosomal recessive inheritance. [1] As a rare disease, Zeichi-Ceide syndrome is registered in the Online Mendelian Inheritance in Man [2] and the U.S. ... S2CID 37739230 . ^ "OMIM Entry - 612916 - ZECHI-CEIDE SYNDROME" . www.omim.org . Retrieved 2016-03-01 . ^ "Zechi Ceide syndrome | Disease | Overview | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov .
Clinical Features Zechi-Ceide et al. (2007) reported 3 sibs, born to consanguineous parents, with occipital atretic cephalocele, hypoplastic cerebellar vermis, Dandy-Walker variant, mental retardation, prominent forehead, short narrow palpebral fissures, midface hypoplasia, broad nose and nasal root, grooved nasal tip and columella, hypoplastic alae nasi with small nares, cleft lip, cleft palate, narrow ears with abnormal helices, antihelices and lobules, narrow auditory canals, oligodontia, large wide feet with a gap between the 1st and 2nd toes, hypoplastic nails, and short metatarsals and distal phalanges. The phenotype was variable in the 3 patients. Inheritance Zechi-Ceide et al. (2007) noted that 3 affected sibs of a consanguineous union strongly suggests autosomal recessive inheritance for this disorder. INHERITANCE - Autosomal recessive HEAD & NECK Head - Prominent forehead Face - Midface hypoplasia Ears - Narrow ears - Narrow auditory canals - Abnormal helices, antihelices and lobules Eyes - Short palpebral fissures - Narrow palpebral fissures Nose - Broad nose - Broad nasal root - Hypoplastic alae nasi - Small nares - Grooved nasal tip Mouth - Cleft lip - Cleft palate - Grooved columella Teeth - Oligodontia SKELETAL Feet - Large wide feet - Gap between the 1st and 2nd toes (sandal gap) - Short metatarsals - Short distal phalanges SKIN, NAILS, & HAIR Nails - Hypoplastic nails NEUROLOGIC Central Nervous System - Occipital atretic cephalocele - Hypoplastic cerebellar vermis - Dandy-Walker variant - Mental retardation ▲ Close
Zechi-Ceide syndrome is a rare, genetic, multiple congenital anomalies/dysmorphic syndrome characterized by occipital atretic cephalocele associated with a specific facial dysmorphism (consisting of prominent forehead, narrow palpebral fissures, midface deficiency, narrow, malformed ears, broad nose and nasal root, grooved nasal tip and columella, laterally angulated, hypoplastic nares, short philtrum, thin upper lip, clift lip/palate, severe oligodontia, prominent chin) and large feet with sandal gap.
Rare syndrome Johnson–Munson syndrome Other names Aphalangy-hemivertebrae-urogenital-intestinal dysgenesis syndrome Aphalangy, hemivertebrae and urogenital-intestinal dysgenesis is an extremely rare syndrome , described only in three siblings. [1] It associates hypoplasia or aplasia of phalanges of hands and feet, hemivertebrae and various urogenital and/or intestinal abnormalities. ... Etiology and inheritance remain unknown. [2] [3] References [ edit ] ^ Johnson VP, Munson DP (Nov 1990). "A new syndrome of aphalangy, hemivertebrae, and urogenital-intestinal dysgenesis". ... PMID 2282714 . ^ Johnson Munson syndrome at NIH 's Office of Rare Diseases ^ Bruno Dallapiccola; Alessandro Castriota-Scanderbeg (2005).
Johnson and Munson (1990) described infant sibs, a female and male, with aphalangy of the hands and feet, hemivertebrae, and visceral malformations which included, in the female, pulmonary hypoplasia, ventricular septal defect, and dysgenesis of the urogenital tract and rectum. The female died in the neonatal period; the male was in good health with normal psychomotor development at the age of 6 months. The mother and her father and grandfather had symphalangism (185800) of both index fingers. Johnson and Munson (1991) reported the birth of another affected brother. Limbs - Aphalangy, hands and feet - Symphalangism of both index fingers in heterozygotes Lungs - Pulmonary hypoplasia Inheritance - Autosomal recessive Spine - Hemivertebrae GU - Dysgenesis of urogenital tract and rectum Cardiac - Ventricular septal defect ▲ Close
An extremely rare congenital limb malformation syndrome, described in only 3 patients to date,characterized by the association of hypoplasia or aplasia of the hand and foot phalanges, hemivertebrae and various urogenital and/or intestinal abnormalities (i.e. dysgenesis of the urogenital tract and rectum).
Saal Bulas syndrome is listed as a "rare disease" by the Office of Rare Diseases (ORD) of the National Institutes of Health (NIH). This means that Saal Bulas syndrome, or a subtype of Saal Bulas syndrome, affects fewer than 200,000 people in the US population. Contents 1 Signs and symptoms 2 Diagnosis 3 Treatment 4 History 5 References 6 External links Signs and symptoms [ edit ] This syndrome consists of ectrodactyly or lobster-like hands, diaphragmatic hernia and absence of the corpus callosum . [1] In addition to these the following problems may also be present. abnormal alimentary tract cardiac septal defect low hair line in front oligodactyly or missing fingers respiratory distress stillbirth/neonatal death Diagnosis [ edit ] This section is empty. ... You can help by adding to it . ( February 2018 ) History [ edit ] The syndrome was first described by American paediatricians Howard M. ... Bulas in 1995. [2] References [ edit ] ^ Bissonnette, Bruno (2006). Syndromes: Rapid Recognition and Perioperative Implications .