-
Burning Mouth Syndrome
Mayo_clinic
The discomfort from burning mouth syndrome can have several different patterns. ... Causes The cause of burning mouth syndrome may be primary or secondary. Primary burning mouth syndrome When the cause can't be found, the condition is called primary or idiopathic burning mouth syndrome. ... Secondary burning mouth syndrome Sometimes burning mouth syndrome is caused by an underlying medical condition. ... Diagnosis There's no one test that can tell if you have burning mouth syndrome. Instead, your health care team will try to rule out other problems before diagnosing burning mouth syndrome. ... Primary burning mouth syndrome There's no known cure for primary burning mouth syndrome.
-
Glaucoma-Ectopia Lentis-Microspherophakia-Stiff Joints-Short Stature Syndrome
Orphanet
Glaucoma-ectopia-microspherophakia-stiff joints-short stature syndrome is characterized by progressive joint stiffness, glaucoma, short stature and lens dislocation. ... The acronym GEMSS (Glaucoma, Ectopia, Microspherophakia, Stiff joints, Short stature) was proposed as a name for the syndrome. This syndrome shows similarities to Moore-Federman syndrome (see this term).
-
Palmoplantar Keratoderma-Deafness Syndrome
Orphanet
Palmoplantar keratoderma-deafness syndrome is a keratinization disorder characterized by focal or diffuse palmoplantar keratoderma. ... Due to genetic and clinical similarities, it has been proposed that palmoplantar keratoderma-deafness syndrome, knuckle pads-leukonychia-sensorineural deafness-palmoplantar hyperkeratosis syndrome and keratoderma hereditarium mutilans may represent variants of one broad disorder of syndromic deafness with heterogeneous phenotype.
-
Gianotti–crosti Syndrome
Wikipedia
Gianotti–Crosti syndrome Specialty Dermatology Gianotti–Crosti syndrome ( / dʒ ə ˈ n ɒ t i ˈ k r ɒ s t i / ), also known as infantile papular acrodermatitis , [1] papular acrodermatitis of childhood , [1] and papulovesicular acrolocated syndrome , [2] : 389 is a reaction of the skin to a viral infection . [3] Hepatitis B virus [4] and Epstein–Barr virus are the most frequently reported pathogens . ... ISBN 0-7216-2921-0 . ^ "Gianotti-crosti syndrome, papulovesicular acrodermatitis. ... (August 2004). "Gianotti-Crosti syndrome caused by acute hepatitis B virus genotype D infection". ... "Diagnostic criteria for Gianotti-Crosti syndrome: a prospective case-control study for validity assessment". ... "The Diagnostic Criteria of Gianotti-Crosti Syndrome: Are They Applicable to Children in India?".
-
Ectrodactyly, Ectodermal Dysplasia, And Cleft Lip/palate Syndrome 1
Omim
This form of ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome, designated EEC1, has been linked to chromosome 7q11.2-q21.3. ... Rodini and Richieri-Costa (1990) reported on 20 Brazilian patients (11 sporadic and 9 familial) and gave a review of 13 disorders to be considered in the differential diagnosis: Rapp-Hodgkin syndrome (129400), the Hay-Wells or AEC syndrome (106260), the Rosselli-Gulienetti syndrome (225000), and others. ... Lacombe et al. (1993) described mother and daughter with features suggesting an overlap between the EEC syndrome and the LADD syndrome (149730). ... Buss et al. (1995) identified 24 cases of EEC syndrome as part of a nationwide study in the UK. ... Scherer et al. (1994) were impressed by the fact that 8 of the patients analyzed were classified as having syndromic ectrodactyly, of which the EEC syndrome is a cardinal example.TP63, PTEN, TP53, CTNNB1, ESR1, UVRAG, CKAP4, PIK3CA, RPE65, CCND1, POLE, PIK3CG, PIK3CB, PIK3CD, RUNX1, KRAS, ETV5, PIK3R1, PGR, MIR429, ARID1A, ERBB2, PCDH10, RASSF1, HIF1A, MALAT1, CDH1, EGFR, AURKA, MIR200C, MIR205, NMU, MAPK8IP2, KDM1A, CCN4, PTENP1, AHSA1, PROM1, CTCF, GRAP2, HAND2, PRDX6, CXCL13, RNF19A, GCA, AKT1, POLDIP2, NEAT1, MIR369, MIR99A, MIR27A, MIR22, MIR206, MIR204, MIR200B, MIR200A, MIR199A2, MIR199A1, MIR181C, MIRLET7C, JAZF1, PDCD4, CD200R1, JPH4, ATG10, SESN2, TET1, ASRGL1, HAND2-AS1, HMGA2, VPS11, DEPDC1, GDE1, JPT1, SLC12A9, PTGER4, AIMP2, HCRTR2, GABPA, MTOR, FOXO1, FOXC1, ESRRG, ENO1, EMD, EIF4A1, DNMT3B, DNMT3A, MAPK14, CRK, CCR1, CHUK, CDKN1A, CD68, CASP3, BCL2, ZFHX3, APC, APBA2, ALCAM, ALB, HCRT, HMGA1, VEGFB, NR4A1, TAF1, STK11, STAT3, SPP1, S100A6, AKT2, MAPK1, PLAU, PIN1, PCNA, PAX2, NME1, NFE2L2, MYC, MRC1, MMP11, SCGB2A1, TACSTD2, LTF, LCT, KDR, IL6, FOXA2, MIR503
-
Cytochrome P450 Oxidoreductase Deficiency
Wikipedia
., CYP3A4 ), [4] among many other CYP450 enzymes. [3] Symptoms of severe forms of PORD include ambiguous genitalia in males and females, congenital adrenal hyperplasia , cortisol deficiency , and Antley–Bixler skeletal malformation syndrome (ABS), while symptoms of mild forms include polycystic ovary syndrome in women and hypogonadism in men. [3] Maternal virilization also occurs in severe forms, due to aromatase deficiency [5] in the placenta . [3] Virilization of female infants in PORD may also be caused by alternative biosynthesis of 5α-dihydrotestosterone via the so-called " androgen backdoor pathway ". [6] The ABS component of severe forms of PORD is probably caused by CYP26B1 deficiency , which results in retinoic acid excess and defects during skeletal embryogenesis . [3] All forms of PORD in humans are likely partial, as POR knockout in mice results in death during prenatal development . [3] See also [ edit ] Androgen backdoor pathway Cytochrome P450 reductase References [ edit ] ^ Flück, Christa E.; Pandey, Amit V. (2019-01-01), "Human P450 Oxidoreductase Deficiency" , in Huhtaniemi, Ilpo; Martini, Luciano (eds.), Encyclopedia of Endocrine Diseases (Second Edition) , Academic Press, pp. 431–443, doi : 10.1016/b978-0-12-801238-3.64966-8 , ISBN 978-0-12-812200-6 , retrieved 2020-02-18 ^ Pandey, Amit V.; Flück, Christa E. ... PMID 31611378 . v t e Gonadal disorder Ovarian Polycystic ovary syndrome Premature ovarian failure Estrogen insensitivity syndrome Hyperthecosis Testicular Enzymatic 5α-reductase deficiency 17β-hydroxysteroid dehydrogenase deficiency aromatase excess syndrome Androgen receptor Androgen insensitivity syndrome Familial male-limited precocious puberty Partial androgen insensitivity syndrome Other Sertoli cell-only syndrome General Hypogonadism Delayed puberty Hypergonadism Precocious puberty Hypoandrogenism Hypoestrogenism Hyperandrogenism Hyperestrogenism Postorgasmic illness syndrome Cytochrome P450 oxidoreductase deficiency Cytochrome b5 deficiency Androgen-dependent condition Aromatase deficiency Complete androgen insensitivity syndrome Mild androgen insensitivity syndrome Hypergonadotropic hypogonadism Hypogonadotropic hypogonadism Fertile eunuch syndrome Estrogen-dependent condition Premature thelarche Gonadotropin insensitivity Hypergonadotropic hypergonadism v t e Inborn errors of steroid metabolism Mevalonate pathway HMG-CoA lyase deficiency Hyper-IgD syndrome Mevalonate kinase deficiency To cholesterol 7-Dehydrocholesterol path: Hydrops-ectopic calcification-moth-eaten skeletal dysplasia CHILD syndrome Conradi-Hünermann syndrome Lathosterolosis Smith–Lemli–Opitz syndrome desmosterol path: Desmosterolosis Steroids Corticosteroid (including CAH ) aldosterone : Glucocorticoid remediable aldosteronism cortisol / cortisone : CAH 17α-hydroxylase CAH 11β-hydroxylase both: CAH 3β-dehydrogenase CAH 21-hydroxylase Apparent mineralocorticoid excess syndrome/11β-dehydrogenase Sex steroid To androgens 17α-Hydroxylase deficiency 17,20-Lyase deficiency Cytochrome b 5 deficiency 3β-Hydroxysteroid dehydrogenase deficiency 17β-Hydroxysteroid dehydrogenase deficiency 5α-Reductase deficiency Pseudovaginal perineoscrotal hypospadias To estrogens Aromatase deficiency Aromatase excess syndrome Other X-linked ichthyosis Antley–Bixler syndrome This article about a disease , disorder, or medical condition is a stub .
-
Adams–stokes Syndrome
Wikipedia
Stokes-Adams syndrome Other names Adams–Stokes syndrome , Gerbezius–Morgagni–Adams–Stokes syndrome and Gerbec–Morgagni–Adams–Stokes syndrome [1] Specialty Cardiology Stokes–Adams syndrome or Adams–Stokes syndrome is a periodic fainting spell in which there is intermittent complete heart block or other high-grade arrhythmia that results in loss of spontaneous circulation and inadequate blood flow to the brain. Subsequently named after two Irish physicians, Robert Adams (1791–1875) [2] and William Stokes (1804–1877), [3] the first description of the syndrome is believed to have been published in 1717 by the Carniolan physician of Slovene descent Marko Gerbec . ... Dallas, TX: FA Davis. p. 903. ^ ADams and victor's principles of neurology ^ Chart 63: Faintness and Fainting , page 161, ISBN 0-86318-864-8 External links [ edit ] Classification D ICD - 10 : I45.9 ICD - 9-CM : 426.9 MeSH : D000219 DiseasesDB : 12443 v t e Cardiovascular disease (heart) Ischaemic Coronary disease Coronary artery disease (CAD) Coronary artery aneurysm Spontaneous coronary artery dissection (SCAD) Coronary thrombosis Coronary vasospasm Myocardial bridge Active ischemia Angina pectoris Prinzmetal's angina Stable angina Acute coronary syndrome Myocardial infarction Unstable angina Sequelae hours Hibernating myocardium Myocardial stunning days Myocardial rupture weeks Aneurysm of heart / Ventricular aneurysm Dressler syndrome Layers Pericardium Pericarditis Acute Chronic / Constrictive Pericardial effusion Cardiac tamponade Hemopericardium Myocardium Myocarditis Chagas disease Cardiomyopathy Dilated Alcoholic Hypertrophic Tachycardia-induced Restrictive Loeffler endocarditis Cardiac amyloidosis Endocardial fibroelastosis Arrhythmogenic right ventricular dysplasia Endocardium / valves Endocarditis infective endocarditis Subacute bacterial endocarditis non-infective endocarditis Libman–Sacks endocarditis Nonbacterial thrombotic endocarditis Valves mitral regurgitation prolapse stenosis aortic stenosis insufficiency tricuspid stenosis insufficiency pulmonary stenosis insufficiency Conduction / arrhythmia Bradycardia Sinus bradycardia Sick sinus syndrome Heart block : Sinoatrial AV 1° 2° 3° Intraventricular Bundle branch block Right Left Left anterior fascicle Left posterior fascicle Bifascicular Trifascicular Adams–Stokes syndrome Tachycardia ( paroxysmal and sinus ) Supraventricular Atrial Multifocal Junctional AV nodal reentrant Junctional ectopic Ventricular Accelerated idioventricular rhythm Catecholaminergic polymorphic Torsades de pointes Premature contraction Atrial Junctional Ventricular Pre-excitation syndrome Lown–Ganong–Levine Wolff–Parkinson–White Flutter / fibrillation Atrial flutter Ventricular flutter Atrial fibrillation Familial Ventricular fibrillation Pacemaker Ectopic pacemaker / Ectopic beat Multifocal atrial tachycardia Pacemaker syndrome Parasystole Wandering atrial pacemaker Long QT syndrome Andersen–Tawil Jervell and Lange-Nielsen Romano–Ward Cardiac arrest Sudden cardiac death Asystole Pulseless electrical activity Sinoatrial arrest Other / ungrouped hexaxial reference system Right axis deviation Left axis deviation QT Short QT syndrome T T wave alternans ST Osborn wave ST elevation ST depression Strain pattern Cardiomegaly Ventricular hypertrophy Left Right / Cor pulmonale Atrial enlargement Left Right Athletic heart syndrome Other Cardiac fibrosis Heart failure Diastolic heart failure Cardiac asthma Rheumatic fever
-
Kocher–debre–semelaigne Syndrome
Wikipedia
Kocher–Debré–Semelaigne syndrome Other names Muscular pseudohypertrophy-hypothyroidism syndrome The Kocher–Debré–Semelaigne syndrome is hypothyroidism in infancy or childhood characterised by lower extremity or generalized muscular hypertrophy, myxoedema , short stature and cretinism . [1] The absence of painful spasms and pseudomyotonia differentiates this syndrome from its adult form, which is Hoffmann syndrome. [2] The syndrome is named after Emil Theodor Kocher , Robert Debré and Georges Semelaigne. Also known as Debré–Semelaigne syndrome or cretinism-muscular hypertrophy, hypothyroid myopathy, hypothyroidism-large muscle syndrome, hypothyreotic muscular hypertrophy in children, infantile myxoedema-muscular hypertrophy, myopathy-myxoedema syndrome, myxoedema-muscular hypertrophy syndrome, myxoedema-myotonic dystrophy syndrome. ... S2CID 11587035 . ^ Rajvanshi, Satyam; Rai, GopalK; Philip, Rajeev; Gupta, KK (2012). "Kocher-Debre-Semelaigne syndrome". Thyroid Research and Practice . 9 (2): 53. doi : 10.4103/0973-0354.96047 . ISSN 0973-0354 . ^ Panat, Sunil R.; Jha, Prakash Chandra; Chinnannavar, Sangamesh N.; Chakarvarty, Ankkita; Aggarwal, Ashish (March 2013). "Kocher Debre Semelaigne Syndrome: A Rare Case Report with Orofacial Manifestations" . ... OCLC 1076268769 . ^ Mehrotra, P (2002-03-01). "Kocher Debre Semelaigne syndrome: regression of pesudohypertrophy of muscles on thyroxine" .
-
Scott Syndrome
Wikipedia
Scott syndrome Other names Platelet factor X receptor deficiency This condition is inherited in an autosomal recessive manner Scott syndrome is a rare congenital bleeding disorder that is due to a defect in a platelet mechanism required for blood coagulation . [1] Normally when a vascular injury occurs, platelets are activated and phosphatidylserine (PS) in the inner leaflet of the platelet membrane is transported to the outer leaflet of the platelet membrane, where it provides a binding site for plasma protein complexes that are involved in the conversion of prothrombin to thrombin , such as factor VIIIa-IXa ( tenase ) and factor Va-Xa ( prothrombinase ). [2] In Scott syndrome, the mechanism for translocating PS to the platelet membrane is defective, resulting in impaired thrombin formation. [3] [4] [5] A similar defect in PS translocation has also been demonstrated in Scott syndrome red blood cells and Epstein-Barr virus transformed lymphocytes , suggesting that the defect in Scott syndrome reflects a mutation in a stem cell that affects multiple hematological lineages. ... References [ edit ] ^ Weiss HJ. Scott syndrome: a disorder of platelet coagulant activity (PCA). Sem Hemat 1994; 31:312-319 ^ Zwaal FA, Comfurius P, Bevers EM. Scott syndrome, a bleeding disorder caused by defective scrambling of membrane phospholipids. ... Characterization of lymphocyte responses to Ca2+ in Scott syndrome. Thromb Haemost 2004; 91:412-415 ^ Sims PJ, Wiedmer T. ... Scott syndrome erythrocytes contain a membrane protein capable of mediating Ca2+-dependent transbilayer migration of membrane phospholipids.

-
Branchio-Oculo-Facial Syndrome
Medlineplus
Branchio-oculo-facial syndrome is a condition that affects development before birth, particularly of structures in the face and neck. ... Problems with branchial arch development underlie many of the other features of branchio-oculo-facial syndrome. "Oculo-" refers to the eyes. Many people with branchio-oculo-facial syndrome have malformations of the eyes that can lead to vision impairment. ... Frequency Branchio-oculo-facial syndrome is a rare condition, although the prevalence is unknown. Causes Branchio-oculo-facial syndrome is caused by mutations in the TFAP2A gene. ... Without this function, transcription factor AP-2α cannot control the activity of genes during development, which disrupts the development of the eyes, ears, and face and causes the features of branchio-oculo-facial syndrome. Learn more about the gene associated with Branchio-oculo-facial syndrome TFAP2A Inheritance Pattern Branchio-oculo-facial syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

-
Desanto-Shinawi Syndrome
Wikipedia
DeSanto-Shinawi syndrome Specialty Neurology , medical genetics Symptoms Specific developmental disorder , intellectual disability , hypotonia , behavioral abnormalities , facial differences, gastrointestinal and eye abnormalities Usual onset Infancy Management Physical , occupational and speech therapy DeSanto-Shinawi ( DESSH ) syndrome is a rare genetic disorder caused by genetic variations (mutations) in a gene called WW Domain-Containing Adaptor with Coiled-coil Region (the WAC gene) . [1] The condition was first described in 2015 in six individuals. [2] The prevalence of DESSH syndrome is unknown at this time, but 25 individuals have been so far described in the medical literature. ... Language acquisition is delayed in almost all individuals with DESSH syndrome. Many patients with DESSH syndrome have cognitive disabilities ranging from mild to moderate. ... Treatment [ edit ] Currently there is no cure for DESSH syndrome and the treatment relies on managing its symptoms. ... fbclid=IwAR301qkC6wih5_p6j_wqMg9P76FzOWru0PYPrO2WefTGwDCruDpZ4uOhxaA ^ a b c d e f "DeSanto-Shinawi Syndrome" . External links [ edit ] [1] DESSH.ORG [2] Dr. Marwan Shinawi MD [3] Facebook Page for Awareness [4] Facebook Family Support Page for DESSH Families ^ "Home | DeSanto Shinawi Syndrome" . DeSanto-Shinawi . Retrieved 2020-03-25 . ^ "Dr.
-
Schwartz-Jampel Syndrome
Medlineplus
Schwartz-Jampel syndrome is a rare condition characterized by permanent muscle stiffness (myotonia) and bone abnormalities known as chondrodysplasia. ... Researchers have since discovered that the condition they thought was Schwartz-Jampel syndrome type 2 is actually part of another disorder, Stüve-Wiedemann syndrome, which is caused by mutations in a different gene. They have recommended that the designation Schwartz-Jampel syndrome type 2 no longer be used. Frequency Schwartz-Jampel syndrome appears to be a rare condition. About 150 cases have been reported in the medical literature. Causes Schwartz-Jampel syndrome is caused by mutations in the HSPG2 gene. ... The mutations that cause Schwartz-Jampel syndrome reduce the amount of perlecan that is produced or lead to a version of perlecan that is only partially functional.
-
Hyperphosphatasia With Mental Retardation Syndrome
Wikipedia
Hyperphosphatasia with mental retardation syndrome Other names image_size = 240px This condition is inherited in an autosomal recessive manner Hyperphosphatasia with mental retardation syndrome , HPMRS , [1] also known as Mabry syndrome , [2] has been described in patients recruited on four continents world-wide. [3] Mabry syndrome was confirmed [4] to represent an autosomal recessive syndrome characterized by severe mental retardation , considerably elevated serum levels of alkaline phosphatase , hypoplastic terminal phalanges, and distinct facial features that include: hypertelorism , a broad nasal bridge and a rectangular face. ... "Hyperphosphatasia with seizures, neurologic deficit, and characteristic facial features: Five new patients with Mabry syndrome". American Journal of Medical Genetics. 152A(7):1661-1669. ^ Thompson MD, Killoran A, Percy ME, Nezarati M, Cole DE, Hwang PA (2006). ... "Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome". Nature Genetics. 42(10):827-9. ^ Thompson MD, Roscioli T, Nezarati MM, Sweeney E, Meinecke P, Krawitz PM, Mabry CC, Horn D, Mendoza R, van Bokhoven H, Stephani F, Marcelis C, Munnich A, Brunner HB, Cole DE (2010). "Heterogeneity of Mabry syndrome: hyperphosphatasia with seizures, neurologic deficit and characteristic facial features". 60 . ... External links [ edit ] Classification D OMIM : 239300 External resources Orphanet : 247262 v t e Inborn error of lipid metabolism : Phospholipid metabolism disorders Tafazzin 3-Methylglutaconic aciduria 2 ( Barth syndrome ) CMD3A Other Paroxysmal nocturnal hemoglobinuria Hyperphosphatasia with mental retardation syndrome
-
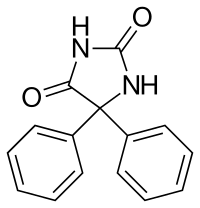
Fetal Hydantoin Syndrome
Wikipedia
Fetal hydantoin syndrome Phenytoin Specialty Medical genetics Fetal hydantoin syndrome , also called fetal dilantin syndrome , is a group of defects caused to the developing fetus by exposure to teratogenic effects of phenytoin . ... It may also be called congenital hydantoin syndrome , [1] fetal hydantoin syndrome , dilantin embryopathy , or phenytoin embryopathy . ... It is important to note that the majority of infants born to women who take phenytoin during pregnancy will not develop fetal hydantoin syndrome. [4] Treatment [ edit ] The treatment of fetal hydantoin syndrome is directed toward the specific symptoms that are apparent in each individual. ... PMID 4644176 . ^ a b c "Fetal Hydantoin Syndrome" . NORD (National Organization for Rare Disorders) . ... External links [ edit ] Classification D ICD - 10 : Q86.1 ICD - 9-CM : 760.77 OMIM : 132810 MeSH : C537922 DiseasesDB : 33179 v t e Congenital malformation due to substance exposure Fetal alcohol spectrum disorder Fetal hydantoin syndrome Fetal warfarin syndrome Prenatal amphetamine exposure Prenatal cannabis exposure Prenatal cocaine exposure Prenatal nicotine exposure Other Substance use disorder
-
Jawad Syndrome
Omim
A number sign (#) is used with this entry because of evidence that Jawad syndrome (microcephaly with mental retardation and digital anomalies) is caused by homozygous mutation in the RBBP8 gene (604124) on chromosome 18q11.2. Seckel syndrome-2 (606744), a microcephaly syndrome involving growth retardation and a characteristic facial appearance, is also caused by mutation in the RBBP8 gene. ... Although the phenotype showed similarities to Filippi syndrome (272440), Kelly et al. (1993) considered it distinct because the patients did not have cryptorchidism, striking physical retardation, or striking facial changes. Hassan et al. (2008) reported a consanguineous Pakistani family with a syndromic form of congenital microcephaly. ... Exclusion Studies In a consanguineous Pakistani family with syndromic microcephaly mapping to chromosome 18p11.22-q11.2, Hassan et al. (2008) analyzed 6 candidate genes but identified no mutations.
-
Asperger Syndrome, Susceptibility To, 1
Omim
Description Asperger syndrome is considered to be a form of childhood autism (see, e.g., 209850). The DSM-IV (American Psychiatric Association, 1994) specifies several diagnostic criteria for Asperger syndrome, which has many of the same features as autism. ... Gillberg et al. (2001) described the development of the Asperger syndrome (and high-functioning autism) Diagnostic Interview (ASDI), which they claimed has a strong validity in the diagnosis of the disorder. Genetic Heterogeneity of Susceptibility to Asperger Syndrome ASPG1 maps to chromosome 3q. Other autosomal loci include ASPG2 (608631) on chromosome 17p, ASPG3 (608781) on 1q21-q22, and ASPG4 (609954) on 3p24-p21. ... In 18 families with both autism and Asperger syndrome, the most significant evidence for linkage was found on chromosome 3q25-q27, with a maximum 2-point lod score of 4.31 at theta = 0.0 with D3S3037.
-
Lig4 Syndrome
Wikipedia
LIG4 syndrome Other names Ligase IV syndrome Person at age 12 showing dysmorphic features LIG4 syndrome (also known as Ligase IV syndrome ) is an extremely rare condition caused by mutations in the DNA Ligase IV (LIG4) gene. ... Some patients have a severe immunodeficiency characterized by pancytopenia , causing chronic respiratory infections and sinusitis. [3] Clinical features also include Seckel syndrome -like facial abnormalities and microcephaly . ... Although not present in all, patients may also present with hypothyroidism and type II diabetes and possibly malignancies such as acute T-cell leukemia . [3] [4] The clinical phenotype of LIG4 syndrome closely resembles that of Nijmegen breakage syndrome (NBS). [ citation needed ] See also [ edit ] LIG4 List of cutaneous conditions References [ edit ] ^ "LIGASE IV, DNA, ATP-DEPENDENT; LIG4" . ... Retrieved 2 January 2012 . ^ Altmann T, Gennery AR (October 2016). "DNA ligase IV syndrome; a review" . Orphanet J Rare Dis . 11 (1): 137. doi : 10.1186/s13023-016-0520-1 . ... "A patient with mutations in DNA Ligase IV: clinical features and overlap with Nijmegen breakage syndrome". Am J Med Genet A . 137A (3): 283–7. doi : 10.1002/ajmg.a.30869 .

-
Nodular Vasculitis
Wikipedia
Most of these cases are now thought to be manifestation of tuberculosis and indeed they respond well to anti-tuberculous treatment. [ citation needed ] See also [ edit ] Panniculitis List of cutaneous conditions References [ edit ] External links [ edit ] Classification D ICD - 10 : L95.8 ( ILDS L95.850) External resources eMedicine : article/1083213 v t e Cutaneous vasculitis and other vascular-related cutaneous conditions Cutaneous vasculitis Erythema elevatum diutinum Capillaritis Urticarial vasculitis Nodular vasculitis Microvascular occlusion Calciphylaxis Cryoglobulinemic purpura / Cryoglobulinemic vasculitis vascular coagulopathy : Livedoid vasculitis Livedoid dermatitis Perinatal gangrene of the buttock Malignant atrophic papulosis Sneddon's syndrome Purpura Nonthrombocytopenic purpura : Cryofibrinogenemic purpura Drug-induced purpura Food-induced purpura IgA vasculitis Obstructive purpura Orthostatic purpura Purpura fulminans Purpura secondary to clotting disorders Purpuric agave dermatitis Pigmentary purpuric eruptions Solar purpura Traumatic purpura Waldenström hyperglobulinemic purpura Painful bruising syndrome ungrouped: Paroxysmal hand hematoma Postcardiotomy syndrome Deep vein thrombosis Superficial thrombophlebitis Mondor's disease Blueberry muffin baby Fibrinolysis syndrome Systemic vasculitis see Template:Systemic vasculitis Vascular malformations Arteriovenous malformation Bonnet–Dechaume–Blanc syndrome Cobb syndrome Parkes Weber syndrome Sinusoidal hemangioma lymphatic malformation Hennekam syndrome Aagenaes syndrome telangiectasia : Generalized essential telangiectasia Hereditary hemorrhagic telangiectasia Unilateral nevoid telangiectasia Ulcer Venous ulcer Arterial insufficiency ulcer Hematopoietic ulcer Neuropathic ulcer Acroangiodermatitis Lymphedema see Template:Lymphatic vessel disease Ungrouped vascular-related cutaneous conditions Raynaud's phenomenon Thromboangiitis obliterans Erythromelalgia Septic thrombophlebitis Arteriosclerosis obliterans Bier spots / Marshall–White syndrome Cholesterol embolus Reactive angioendotheliomatosis Trousseau's syndrome v t e Disorders of subcutaneous fat Panniculitis Lobular without vasculitis Cold Cytophagic histiocytic Factitial Gouty Pancreatic Traumatic needle-shaped clefts Subcutaneous fat necrosis of the newborn Sclerema neonatorum Post-steroid panniculitis Lipodermatosclerosis Weber–Christian disease Lupus erythematosus panniculitis Sclerosing lipogranuloma with vasculitis: Nodular vasculitis / Erythema induratum Septal without vasculitis: Alpha-1 antitrypsin deficiency panniculitis Erythema nodosum Acute Chronic with vasculitis: Superficial thrombophlebitis Lipodystrophy Acquired generalized: Acquired generalized lipodystrophy partial: Acquired partial lipodystrophy Centrifugal abdominal lipodystrophy HIV-associated lipodystrophy Lipoatrophia annularis localized: Localized lipodystrophy Congenital Congenital generalized lipodystrophy Familial partial lipodystrophy Marfanoid–progeroid–lipodystrophy syndrome Poland syndrome This cutaneous condition article is a stub .
-
Alpers-Huttenlocher Syndrome
Medlineplus
Alpers-Huttenlocher syndrome is one of the most severe of a group of conditions called the POLG -related disorders. ... People with Alpers-Huttenlocher syndrome usually have additional signs and symptoms. ... Some people with Alpers-Huttenlocher syndrome lose the ability to walk, sit, or feed themselves. ... Frequency The prevalence of Alpers-Huttenlocher syndrome is approximately 1 in 100,000 individuals. Causes Alpers-Huttenlocher syndrome is caused by mutations in the POLG gene.POLG, PARS2, RLBP1, FANCI, EDAR, AHSG, ABL2, NARS2, TWNK, DARS2, FARS2, SOD1, SNCB, SMN2, SMN1, PRNP, NPTX2, MPV17, IGFALS, HTC2, HTT, TYMP, DGUOK, CTSC, CARS1, RERE, FAS, OSR1

-
Burn-Mckeown Syndrome
Medlineplus
Burn-McKeown syndrome is a disorder that is present from birth (congenital) and involves abnormalities of the nasal passages, characteristic facial features, hearing loss, heart abnormalities, and short stature. ... Other features that can occur in Burn-McKeown syndrome include mild short stature and congenital heart defects such as patent ductus arteriosus (PDA). ... Intelligence is unaffected in Burn-McKeown syndrome. Frequency Burn-McKeown syndrome is a rare disorder; its prevalence is unknown. ... The mutations affecting the TXNL4A gene that cause Burn-McKeown syndrome reduce the amount of protein produced from the gene. ... Learn more about the gene associated with Burn-McKeown syndrome TXNL4A Inheritance Pattern This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations.




