-
Hypospadias 3, Autosomal
Omim
Inheritance Lowry and Kliman (1976) suggested, on the basis of 2 kindreds, that an autosomal dominant form may account for a small number of hypospadias cases. Cote et al. (1979) described 4 males with hypospadias of the midshaft of the penis in 3 generations, with 1 male-to-male transmission and transmission through a female. Page (1979) also suggested autosomal dominant inheritance for some cases. ... Mapping Thai et al. (2008) performed genomewide linkage analysis of a 3-generation family of Finnish origin in which 7 individuals had isolated hypospadias inherited in an autosomal dominant pattern. A maximum parametric lod score of 2.71 was found at marker D7S640 on chromosome 7q32.2-q36.1 (nonparametric lod score of 5.01). ... Mutation analysis excluded pathogenic mutations in the AKR1D1 (604741) and PTN (162095) genes. INHERITANCE - Autosomal dominant - Multifactorial GENITOURINARY External Genitalia (Male) - Hypospadias MISCELLANEOUS - See also two X-linked forms 300633 and 300758 ▲ Close
-
First Arch Syndrome
Wikipedia
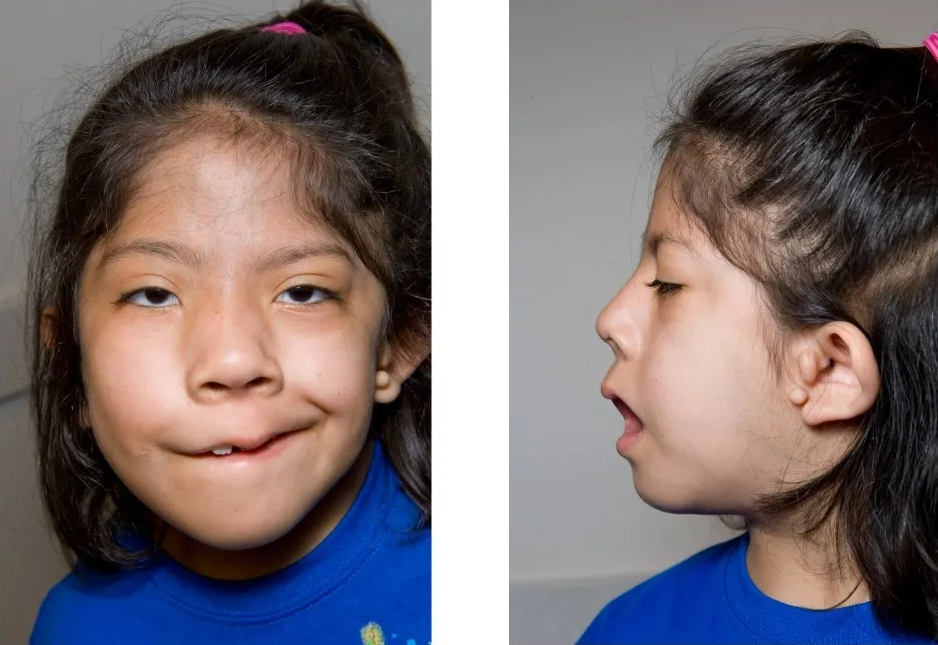
First arch syndrome Specialty Medical genetics First arch syndromes are congenital defects caused by a failure of neural crest cells to migrate into the first pharyngeal arch . [1] They can produce facial anomalies . Examples of first arch syndromes include Treacher Collins syndrome and Pierre Robin syndrome . ... Dudek. High-Yield Embryology. 2e. Page 65. This article about a congenital malformation is a stub .MYT1, TBC1D20, RAB18, ZIC3, NID2, PARD3B, AGAP1, SHROOM3, FRMD4A, ARID3B, PART1, SALL1, OTX2, FGF8, EFTUD2, TSC1, CHD7, SMARCB1, SIX1, WNT5B, COX8A
-
Tooth Agenesis, Selective, 3
Omim
Frazier-Bowers et al. (2002) described a large family with autosomal dominant oligodontia. Affected individuals showed predominantly molar oligodontia but it was not limited to posterior teeth. ... Several second premolars were missing, even though the neighboring first molars were present. Mostowska et al. (2006) described a 3-generation family with severe autosomal dominant oligodontia. ... Inheritance Selective tooth agenesis-3 is an autosomal dominant anomaly (Stockton et al., 2000; Frazier-Bowers et al., 2002). ... Frazier-Bowers et al. (2002) identified a novel insertion mutation in the PAX9 gene (167416.0010) in a large family with autosomal dominant oligodontia. Lammi et al. (2003) identified a novel missense mutation in the PAX9 gene (167416.0008) in affected members of a Finnish family segregating autosomal dominant oligodontia. ... MSX1-associated tooth agenesis (STHAG1; 106600) typically includes missing maxillary and mandibular second premolars and maxillary first premolars. The most distinguishing feature of MSX1-associated tooth agenesis is the frequent (75%) absence of maxillary first premolars, whereas the most distinguishing feature of PAX9-associated tooth agenesis is the frequent (over 80%) absence of maxillary and mandibular second molars.
-
Aortic Aneurysm, Familial Thoracic 10
Omim
Clinical Features Guo et al. (2016) studied 6 families segregating autosomal dominant thoracic aortic aneurysm with or without dissection. ... Lee et al. (2016) reported a family segregating autosomal dominant thoracic aortic aneurysm, in which the proband was a 39-year-old man who had a 10.5-cm ascending aortic aneurysm with contained rupture at age 19 years. ... Molecular Genetics In 3 patients from a family segregating autosomal dominant thoracic aortic aneurysm with or without dissection, who were negative for mutation in known thoracic aortic aneurysm-associated genes, Guo et al. (2016) performed whole-exome sequencing and identified heterozygosity for a missense mutation in the LOX gene (S280R; 153455.0002) that segregated with disease in the family. ... Guo et al. (2016) concluded that LOX variants are responsible for approximately 1% of familial thoracic aortic disease and are associated with decreased penetrance. In 2 first cousins with thoracic aortic aneurysm and dissection or rupture, one of whom exhibited features of Marfan syndrome but was negative for mutation in the FBN1 (134797), TGFBR1 (190181), and TGFBR2 (190182) genes, Lee et al. (2016) performed whole-genome sequencing and identified heterozygosity for a missense mutation in the LOX gene (M298R; 153455.0005). The mutation was also detected in the proband's affected mother and in his cousin's affected brother, as well as in the cousins' mother, who exhibited only age-related arterial tortuosity. INHERITANCE - Autosomal dominant GROWTH Height - Tall stature (in some patients) HEAD & NECK Eyes - Myopia (rare) Mouth - High-arched palate (in some patients) Teeth - Dental crowding (rare) CARDIOVASCULAR Heart - Bicuspid aortic valve (in some patients) - Mitral regurgitation (rare) - Coronary artery disease (rare) Vascular - Aortic root aneurysm - Aneurysm of ascending aorta - Fusiform aneurysm involving aortic root and ascending aorta - Dissection and/or rupture of aortic aneurysm (in some patients) - Aortic arch aneurysm (rare) - Abdominal aortic aneurysm (rare) - Hepatic artery aneurysm (rare) - Venous varicosities of the lower extremities (rare) CHEST Ribs Sternum Clavicles & Scapulae - Pectus excavatum (in some patients) ABDOMEN External Features - Abdominal hernia (rare) SKELETAL Spine - Scoliosis (in some patients) Limbs - Joint hypermobility (in some patients) - Dolichostenomelia (rare) SKIN, NAILS, & HAIR Skin - Skin striae (in some patients) NEUROLOGIC Central Nervous System - Dural ectasia (in some patients) MOLECULAR BASIS - Caused by mutation in the lysyl oxidase gene (LOX, 153455.0002 ) ▲ Close
-
Rosah Syndrome
Wikipedia
Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This article includes a list of general references , but it remains largely unverified because it lacks sufficient corresponding inline citations . ... Find sources: "ROSAH syndrome" – news · newspapers · books · scholar · JSTOR ( June 2019 ) ( Learn how and when to remove this template message ) ( Learn how and when to remove this template message ) ROSAH syndrome ROSAH syndrome is inherited via an autosomal dominant manner Causes Mutation in ALPK1 gene ROSAH syndrome is a rare genetic disorder characterised by Retinal dystrophy, Optic nerve edema, Splenomegaly, Anhidrosis and migraine Headache. [1] Contents 1 Presentation 2 Genetics 3 Diagnosis 4 Management 5 Epidemiology 6 History 7 References Presentation [ edit ] The main characteristics of this condition are retinal dystrophy, optic nerve oedema, splenomegaly , anhidrosis and migraine headache. ... The inheritance of this condition is autosomal dominant. Diagnosis [ edit ] This diagnosis is made by sequencing the ALPK1 gene. ... History [ edit ] This condition was first described in 2019. [1] References [ edit ] ^ a b Williams LB, Javed A, Sabri A, Morgan DJ, Huff CD, Grigg JR, Heng XT, Khng AJ, Hollink IHIM, Morrison MA, Owen LA, Anderson K, Kinard K, Greenlees R, Novacic D, Nida Sen H, Zein WM, Rodgers GM, Vitale AT, Haider NB, Hillmer AM, Ng PC, Shankaracharya, Cheng A, Zheng L, Gillies MC, van Slegtenhorst M, van Hagen PM, Missotten TOAR, Farley GL, Polo M, Malatack J, Curtin J, Martin F, Arbuckle S, Alexander SI, Chircop M, Davila S, Digre KB, Jamieson RV, DeAngelis MM (2019) ALPK1 missense pathogenic variant in five families leads to ROSAH syndrome, an ocular multisystem autosomal dominant disorder.

-
Visual Agnosia
Wikipedia
Individuals with apperceptive visual agnosia cannot form a whole percept of visual information. [15] Associative visual agnosia , impaired object identification. ... ISBN 978-0-205-66627-0 . OCLC 263605380 . [ page needed ] ^ Goodale MA, Milner AD, Jakobson LS, Carey DP (1991). ... New York, NY., Worth Publishers . ISBN 978-0-7167-9586-5 . [ page needed ] ^ McCarthy, R. A.; Warrington, E. ... New York, NY., Oxford University Press . ISBN 978-0-19-514490-1 . [ page needed ] ^ Biran, I.; Coslett, H. ... "Sensation & Perception" 3rd ed. pp. 507 ISBN 978-0-87893-876-6 . [ page needed ] ^ Harris, Irina M.; Harris, Justin A.; Caine, Diana (2001).
-
Autosomal Dominant Retinal Vasculopathy With Cerebral Leukodystrophy
Wikipedia
Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This article needs additional citations for verification . ... Unsourced material may be challenged and removed. Find sources: "Autosomal dominant retinal vasculopathy with cerebral leukodystrophy" – news · newspapers · books · scholar · JSTOR ( June 2016 ) ( Learn how and when to remove this template message ) This article needs editing for compliance with Wikipedia's Manual of Style . ... Relevant discussion may be found on the talk page . Please help to ensure that disputed statements are reliably sourced . ( August 2015 ) ( Learn how and when to remove this template message ) ( Learn how and when to remove this template message ) Autosomal dominant retinal vasculopathy with cerebral leukodystrophy Other names Retinal vasculopathy and cerebral leukoencephalopathy Diagram depicts the mode of inheritance of this condition Autosomal Dominant Retinal Vasculopathy with Cerebral Leukodystrophy (AD-RVCL) (previously known also as Cerebroretinal Vasculopathy, CRV, or Hereditary Vascular Retinopathy, HVR or Hereditary Endotheliopathy, Retinopathy, Nephropathy, and Stroke, HERNS) is an inherited condition resulting from a frameshift mutation to the TREX1 gene. ... The overall prognosis is poor, and death can sometimes occur within 10 years of the first symptoms appearing. [1] Contents 1 Presentation 1.1 Clinical Associations 2 Genetics 2.1 The immune system 2.1.1 immune system becomes part of the condition 3 Pathogenesis 4 Diagnosis 4.1 Differential diagnosis 5 Treatment 6 History 7 References 8 External links Presentation [ edit ] No recognizable symptoms until after age 40. ... Louis, MO 1988: 10 families worldwide were identified as having CRV 1991: Related disease reported, HERNS (Hereditary Endiotheliopathy with Retinopathy, Nephropathy and Stroke – UCLA 1998: Related disease reported, HRV (Hereditary Retinal Vasculopathy) – Leiden University, Netherlands 2001: Localized to Chromosome 3. 2007: The specific genetic defect in all of these families was discovered in a single gene called TREX1 2008: Name changed to AD-RVCL Autosomal Dominant-Retinal Vasculopathy with Cerebral Leukodystrophy 2009: Testing for the disease available to persons 21 and older 2011: 20 families worldwide were identified as having CRV 2012: Obtained mouse models for further research and to test therapeutic agents References [ edit ] ^ "Retinal vasculopathy with cerebral leukodystrophy | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" .
-
Star Syndrome
Wikipedia
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( September 2017 ) STAR syndrome Other names Syndactyly with Renal and Anogenital Malformation STAR syndrome is a rare X-linked dominant disorder.
-
Autoimmune Lymphoproliferative Syndrome, Type V
Omim
Description Autoimmune lymphoproliferative syndrome type V is an autosomal dominant complex immune disorder characterized by autoimmune thrombocytopenias and abnormal lymphocytic infiltration of nonlymphoid organs, including the lungs, brain, and gastrointestinal tract, resulting in enteropathy. ... The age at onset and presentation was variable, but most patients developed symptoms between late in the first decade and the third decade. Seven patients fulfilled the diagnosis of common variable immunodeficiency (CVID). ... Inheritance The transmission pattern of ALPS5 in the families reported by Kuehn et al. (2014) and Schubert et al. (2014) was consistent with autosomal dominant inheritance and incomplete penetrance. Molecular Genetics In 6 patients from 4 families with autoimmune lymphoproliferative syndrome type V, Kuehn et al. (2014) identified 4 different heterozygous loss-of-function mutations in the CTLA4 gene (see, e.g., 123890.0003-123890.0005). The mutation in the first family was found by whole-exome sequencing; mutations in subsequent families were found by direct screening of the CTLA4 gene in 23 unrelated probands with a similar phenotype. ... In 11 patients from 6 unrelated families with ALPS5, Schubert et al. (2014) identified 6 different heterozygous mutations in the CTLA4 gene (see, e.g., 123890.0006-123890.0008). The mutation in the first family was found by whole-exome sequencing; mutations in subsequent families were found by direct screening of the CTLA4 gene in 71 unrelated probands with CVID and enteropathy or autoimmunity.
-
Tremor, Hereditary Essential, 5
Omim
Description Hereditary essential tremor-5 is an autosomal dominant neurologic disorder characterized by kinetic, intention, and/or postural tremor mainly affecting the upper limbs. ... Inheritance The transmission pattern of ETM5 in the families reported by Hor et al. (2015) was consistent with autosomal dominant inheritance. There was some evidence of age-dependent and/or incomplete penetrance. Molecular Genetics In affected members of 3 unrelated families of Spanish descent with ETM5, Hor et al. (2015) identified 3 different heterozygous missense mutations in the TENM4 gene (610084.0001-610084.0003). The mutation in the first family was found by whole-exome sequencing and confirmed by Sanger sequencing; mutations in the subsequent 2 families were found by targeted resequencing of TENM4 in 299 Spanish probands with essential tremor. ... Injection of the mutations into zebrafish embryos resulted in a defect in axonal guidance similar to that observed with morpholino knockdown of the tenm4 gene and consistent with a dominant-negative effect. Animal Model Suzuki et al. (2012) found that the homozygous mouse mutant, 'furue' (fur), characterized by tremors and hypomyelination in the central nervous system, resulted from a homozygous insertion mutation in the Tenm4 gene. ... These defects could be rescued by wildtype human TENM4. INHERITANCE - Autosomal dominant HEAD & NECK Mouth - Tongue tremor (in some patients) NEUROLOGIC Central Nervous System - Tremor, essential - Postural tremor - Kinetic tremor - Intention tremor - Tremor mainly affects the upper limbs - Tremor may affect the lower limbs MISCELLANEOUS - Variable age at onset (range adolescence to late adulthood) - Variable severity - Slow progression - Age-dependent penetrance - Incomplete penetrance MOLECULAR BASIS - Caused by mutation in the teneurin transmembrane protein 4 gene (TENM4, 610084.0001 ) ▲ Close
-
Reticulate Acropigmentation Of Kitamura
Omim
Description Reticulate acropigmentation of Kitamura (RAK) is a rare pigmentary disorder that usually shows an autosomal dominant pattern of inheritance with high penetrance. Typical features include reticulate, slightly depressed, sharply demarcated brown macules without hypopigmentation, affecting the dorsa of the hands and feet in the first or second decade of life. The macules gradually darken and extend to the proximal regions of the extremities; progression of the eruptions stops in middle age. ... For a discussion of genetic heterogeneity of reticulate pigment disorders, see 179850. Molecular Genetics By whole-exome sequencing in a 4-generation Japanese family with autosomal dominant reticulate acropigmentation, Kono et al. (2013) identified a 5-bp insertion in the ADAM10 gene (602192.0001) that segregated with disease in the family and was not found in 102 Japanese controls. ... KRT5 (148040) mutations were excluded in all of the patients. INHERITANCE - Autosomal dominant SKIN, NAILS, & HAIR Skin - Reticulate hyperpigmented macules which darken over time and appear initially on dorsa of hands and feet but progress to flexor aspects of proximal extremities - Breaks in epidermal ridges of palms and fingers - Palmoplantar pits - Plantar keratoderma (in some patients) Skin Histology - Pigmentation in tip of rete ridges - Epidermal thinning - Elongation and thinning of rete ridges - Slight hyperkeratosis - Few inflammatory cell infiltrates Hair - Partial alopecia MISCELLANEOUS - Onset in first or second decade of life MOLECULAR BASIS - Caused by mutation in the gene encoding a disintegrin and metalloproteinase domain-10 (ADAM10, 602192.0001 ) ▲ Close
-
Cataract 33, Multiple Types
Omim
Description Mutations in the BFSP1 gene have been found to cause multiple types of cataract, which have been described as cortical, nuclear, and progressive punctate lamellar. Both autosomal dominant and autosomal recessive modes of inheritance have been reported. Clinical Features Ramachandran et al. (2007) examined 11 affected and 8 unaffected members of a large consanguineous Indian family (family 30023) segregating autosomal recessive juvenile-onset cortical cataract. Cataract was first documented at 5 years of age in all affected individuals but 1, in whom it was noted at 2 years of age. ... Wang et al. (2013) reported a 5-generation Chinese family in which 15 members had autosomal dominant bilateral nuclear cataract. Opacities were visible in childhood. ... The proband's affected brother and his father were not available for study. Autosomal Dominant Inheritance In a 5-generation Chinese family in which 15 members had autosomal dominant nuclear cataract unlinked to known cataract loci, Wang et al. (2013) performed whole-exome sequencing and identified heterozygosity for a missense mutation (D348N; 603307.0002) in the BFSP1 gene. ... INHERITANCE - Autosomal recessive - Autosomal dominant HEAD & NECK Eyes - Cortical cataract - Nuclear cataract - Punctate lamellar cataract - Cysts in anterior cortex MISCELLANEOUS - Cataracts first documented in early childhood MOLECULAR BASIS - Caused by mutation in the beaded filament structural protein 1 (BFSP1, 603307.0001 ) ▲ CloseCRYAA, GJA8, BFSP1, CRYBB1, WFS1, MIP, CRYBB3, CRYGC, NHS, CRYGD, CRYBB2, CRYBA1, EPHA2, CRYAB, CRYBA2, FYCO1, GJA3, UNC45B, CHMP4B, SCAPER, HSF4, CFAP410, FTL, LMNA, ERCC6, MIR335, MIR34A, RNF149, ELANE, GSTO2, FTO, IRF1, PRDX6, WRN, GSTP1, TGM2, SLC11A2, BLM, MIR630

-
Epileptic Encephalopathy, Early Infantile, 66
Omim
Most patients had onset of seizures in the first days or weeks of life, although 1 had onset at 2 months of age. ... The seizures tended to attenuate with time, especially after the first year of life, and a few patients even became seizure free with treatment. ... Molecular Genetics In 14 unrelated patients with EIEE66, Olson et al. (2018) identified a de novo heterozygous missense mutation in the PACS2 gene (E209K; 610423.0001). The mutation in the first 2 patients was found by whole-exome sequencing and confirmed by Sanger sequencing. ... The findings may be consistent with a dominant-negative effect. INHERITANCE - Autosomal dominant HEAD & NECK Face - Coarse facial features Eyes - Nystagmus - Hypertelorism - Downslanting palpebral fissures - Myopia - Hypermetropia - Strabismus - Astigmatism - Synophrys Nose - Broad nasal root Mouth - Thin upper lip - Wide mouth - Downturned corners of mouth CARDIOVASCULAR Heart - Septal defects (in some patients) GENITOURINARY External Genitalia (Male) - Cryptorchidism SKELETAL Hands - Fifth finger clinodactyly MUSCLE, SOFT TISSUES - Hypotonia NEUROLOGIC Central Nervous System - Epileptic encephalopathy - Seizures, multiple types - Focal and multifocal spike abnormalities seen on EEG - Delayed psychomotor development - Delayed walking - Wide-based gait - Intellectual disability - Speech delay - Mega cisterna magna - Inferior vermian hypoplasia - Cerebellar dysgenesis - Hypothalamic fusion Behavioral Psychiatric Manifestations - Autism spectrum disorder - Stereotypies HEMATOLOGY - Anemia (in some patients) - Neutropenia (in some patients) MISCELLANEOUS - Onset of seizures in first days or weeks of life - Seizures may attenuate somewhat after the first year of life - Variable extraneurologic features - De novo mutation MOLECULAR BASIS - Caused by mutation in the phosphofurin acidic cluster sorting protein 2 gene (PACS2, 610423.0001 ) ▲ Close
-
Lymphatic Malformation 7
Omim
Description LMPHM7 is an autosomal dominant disorder with variable expressivity. ... Clinical Features Martin-Almedina et al. (2016) reported 2 unrelated families from the United Kingdom and Norway, respectively, with an inherited disorder characterized mainly by LRHF and/or ASD. The first family had 7 affected members spanning 3 generations. ... Inheritance The transmission pattern of LMPHM7 in the families reported by Martin-Almedina et al. (2016) was consistent with autosomal dominant inheritance with variable expressivity. ... The mutations, which were found by whole-exome sequencing and confirmed by Sanger sequencing, segregated with the disorder in the families, although there was variable expressivity of the phenotype. ... The findings indicated that the mutations had a negative effect on receptor activity after ligand stimulation. There was no evidence of a dominant-negative effect. In a large 4-generation family with hydrops fetalis, lymphedema, and central conduction lymphatic anomalies, Li et al. (2018) performed whole-exome sequencing and identified heterozygosity for a splice site mutation in the EPHB4 gene (600011.0013) that segregated fully with disease in the family and was not found in in-house control samples or in public variant databases.
-
Hiv/aids In Tanzania
Wikipedia
Refer to page 202 of the survey. ^ At a 95 percent confidence level , the rate was 5.5 to 6.8 percent. Refer to page 202 of the survey. ^ At a 95 percent confidence level , the rate was 3.2 to 4.5 percent. Refer to page 202 of the survey. ^ At a 95 percent confidence level , the rate was 10.8 to 19.9 percent. ... Refer to page 221 of the survey. ^ At a 95 percent confidence level , the rate was 2.0 to 3.3 percent. Refer to page 202 of the survey. ^ At a 95 percent confidence level , the rate was 0.7 to 1.7 percent.

-
Pneumatosis
Wikipedia
A fourth type known as irregular emphysema involves the acinus irregularly and is associated with fibrosis. [11] Though the subtypes can be seen on imaging they are not well-defined clinically. [12] Only the first two types of emphysema – centrilobular, and panlobular are associated with significant airflow obstruction, with that of centrilobular emphysema around 20 times more common than panlobular. [11] Centrilobular emphysema [ edit ] Centrilobular emphysema also called centriacinar emphysema , affects the centrilobular portion of the lung, the area around the terminal bronchiole, and the first respiratory bronchiole, and can be seen on imaging as an area around the tip of the visible pulmonary artery. ... It is diagnosed around the time of birth or in the first 6 months of life, occurring more often in boys than girls. ... In rare cases air may escape from the gland and give rise to subcutaneous emphysema in the face, neck, or mediastinum. [35] [36] Terminology [ edit ] The term pneumatosis has word roots of pneumat- + -osis , meaning "air problem/injury". References [ edit ] ^ page 64 in: Adrian Shifren (2006). The Washington Manual Pulmonary Medicine Subspecialty Consult, Washington manual subspecialty consult series . ... Radiopaedia . Retrieved 2018-01-03 . ^ a b c Page 60 in: Harry Griffiths (2008). Musculoskeletal Radiology . ... ISBN 9781420020663 . ^ Restrepo, Carlos S.; Martinez, Santiago; Lemos, Diego F.; Washington, Lacey; McAdams, H. Page; Vargas, Daniel; Lemos, Julio A.; Carrillo, Jorge A.; Diethelm, Lisa (2009).

-
Dementia Praecox
Wikipedia
Swiss-émigré Adolf Meyer (1866–1950), arguably the most influential psychiatrist in America for the first half of the 20th century, published the first critique of dementia praecox in an 1896 book review of the 5th edition of Kraepelin's textbook. ... Adolf Meyer was the first to apply the new diagnostic term in America. ... It is first mentioned in The New York Times in 1925. ... Dowbiggin inaccurately states that Morel used the term on page 234 of the first volume of his 1852 publication Etudes cliniques ( Dowbiggin 1996 , p. 388; Morel 1852 , p. 234 ). ... In the first instance the reference is made in relation to young girls of asthenic build who have often also suffered from typhoid.AKT1, MAGI2, CHRNA7, SHANK3, TCF4, NOS1, RELN, NRXN1, HTR2A, CSMD1, SRR, GRIN2B, RTN4R, SP4, SETD1A, PPP3R1, SYNGAP1, MDK, NR4A2, KMO, APOE, DTNBP1, GRM5, ZDHHC8, GSK3B, NRG3, DISC1, PPP1R1B, SLC6A3, CNR1, MBP, BACE1, CPLX2, TAAR1, MAP6, PTGS2, YWHAH, PLCB1, LRRTM1, SLC6A1, MAP2K7, NLGN2, MTOR, ZIC2, AVPR1A, GNAS, BECN1, NOS1AP, PDE4B, FEZ1, CHI3L1, DRD3, CACNA1C, ABCB1, GRM3, GRIA1, NRG1, HLA-DRB1, NRGN, PRODH, ANK3, TBX1, SLC1A1, SRGAP3, NOTCH4, MTHFR, BRD1, DISC2, FOXP2, NCAM1, DAOA, COMT, ZNF804A, SYN2, MED12, CHRNA5, VRK2, HLA-DQB1, HLA-A, NDE1, TCF7L2, GABBR1, DLG2, MIR137HG, ARVCF, TSPAN18, CNNM2, YWHAE, FYN, MAD1L1, TSNARE1, MPC2, ITIH3, NTRK3, PBRM1, TNXB, OPCML, DDR1, HLA-B, UFD1, ATP2A2, BCL9, MSRA, NKAPL, CACNB2, PGBD1, ESR2, DCC, TNIK, CACNA1B, NLGN1, RAI1, TENM4, FGFR1, DOCK4, ZKSCAN4, ALDH1A2, ITIH1, BCL11A, GPM6A, AMBRA1, ABCA13, PPP2R2B, PDE4D, CHRNA3, PLAA, CEACAM21, EML5, RTKN2, CTNNA2, PSD3, GJA5, GJA8, SDCCAG8, CYP26B1, ZSCAN31, DPYD, SOX5, TMEM245, MEGF10, HHAT, CDC25C, HFE, PCDH17, APOL2, LRRK2, ADAMTSL3, SLC25A12, GDNF, SLC6A9, DAOA-AS1, PRL, CHRNA4, CCK, FMR1, KLF12, PLA2G4A, GABRB2, CYP1A2, SLC18A2, PPP3CC, ANKK1, PIP4K2A, GLUL, CNP, CYP2D6, DAO, MARK2, CHRM1, GRIN2A, PLA2G1B, GAD1, NR3C1, SST, CHRFAM7A, GRM2, GRM7, ACE, CRP, FZD3, APOL4, EGF, DNMT1, VPS35, NTF3, PSD, HTR1A, KCNN3, HTR2C, PLCL2, DRD1, HTR6, DRD2, IFNG, OLIG2, NPY, DRD4, ST8SIA2, IL1A, CCL2, IL1B, IL2, SYN3, AHI1, IL4, IL6, HOMER1, IL10, PLA2G6, MMP9, NPAS3, FHIT, SLC6A4, MIR137, MAOB, TNF, MAOA, CNTNAP2, TPH2, MAG, ERBB4, TPH1, RTN4, ESR1, BDNF, C9orf72, PVALB, DLG4, NDEL1, ADRA1A, SLC1A2, F2, EGR3, S100B, SOD2, LEP, ADCYAP1, SLC17A7, ACTB, GNAO1, HPGDS, MLC1, CNTF, GFAP, ERBB3, NDUFV2, CLDN5, CCKAR, GSTM1, TP53, VIPR2, DLG1, OXTR, CYFIP1, SOX10, GSTT1, PCM1, TSNAX, GAD2, HLA-C, PDLIM5, VEGFA, NTRK2, HSPA1A, AVP, HTR3A, PIK3CA, IL3, TLR4, SLC1A3, ARC, GABRB3, SLC12A2, CMYA5, NTNG1, SLC18A1, MAP2, HTR1F, KCNH2, SYP, PIK3CB, GRIK3, HTR7, CLOCK, PAH, HINT1, QKI, NTS, MOG, CREB1, ADORA2A, CALM1, SLC12A5, ERVW-1, PLXNA2, NTRK1, HDAC1, PCNT, DLG3, OPRM1, VGF, HDAC2, SOD1, XBP1, BAG1, HSPA1B, GSTP1, HTR5A, PIP4K2C, PICK1, FABP7, IL18, EGR1, NUFIP2, HDAC3, ADNP, SIGMAR1, GRM8, GRID1, CAMKK2, GRM1, CHL1, SREBF1, GRIA2, CTNNB1, CHRM4, GRIK1, APOD, CACNG2, GNB1L, NEFL, AR, HTR4, UHMK1, NCAN, CPLX1, FXYD6, AGA, HTR3B, PPARA, CSF2, MET, SYNGR1, SLC17A6, PON1, LTA, GAP43, SELENBP1, PLP1, GABRA1, SULT4A1, NQO1, PDE4A, PIK3C3, CDC42, SLC6A2, MIR346, DDC, DGCR2, GRIK4, NTNG2, GRIK2, HMOX1, HP, GRIA4, SMARCA2, SP1, PTPN5, MAPK1, KIF17, GRIN2C, PDE7B, MAPK3, GRIN2D, PSEN2, PTPRZ1, HK1, GCLM, TET1, GABRG2, GLS, IL6R, NRN1, GRIN3A, MCHR1, GPX1, HSPA1L, PINK1, IPO5, NCS1, ACSL6, EGR2, TYR, MIR30E, TNFRSF1A, DNM1, SLC1A4, SIRPB1, TGFB1, TF, CHGB, CYP3A4, CNR2, SREBF2, CSF2RA, CTLA4, ASTN2, MYT1L, NDST3, DLGAP1, ADCYAP1R1, ADRA2A, NRXN3, TBP, CTCF, NPSR1, ADM, TGM2, CHAT, GSTM2, KPNA3, PDYN, DDO, KCNQ2, GRN, PLA2G4C, PI4KA, TLR3, CLU, GNAL, KMT2A, CCR5, GLO1, PPARGC1A, RGS9, B3GAT1, CRYZ, MAPK14, ERDA1, PHOX2B, PPP1R9B, CALB1, ULK4, JUN, RCBTB1, IL10RA, ERVK-18, MYO9B, IL3RA, DGCR6, CARTPT, AQP4, ICAM1, HDAC4, XRCC1, LINC00271, CCND1, ARHGEF11, VSNL1, RAPGEF6, VIP, OPA1, VDR, MDGA1, OXT, PAFAH1B1, PAX6, TYROBP, MTNR1B, ACTR2, TNFRSF1B, CSF2RB, RHD, MIR206, SLC1A5, SIRT1, EFNB2, EGR4, CYP2E1, PTGS1, SLC6A12, ELAVL2, LHX6, SLC32A1, RARA, SLC1A6, UCP2, MIR432, PAK2, HLA-DPB1, RANBP9, PAWR, CTXN3, MTNR1A, NPS, SCGB1A1, SRSF1, OTX2, OPRK1, MED15, PTPN1, AGER, ETNPPL, SHANK1, NR2E1, DCLK1, STON2, PRSS16, EPHX2, PDGFRB, SLC25A27, ADRB3, FGFR2, CAMK2B, TTR, MIR17, RGS10, KCNS3, HCRTR1, DLGAP2, DNMT3B, VLDLR, MTHFD1, CD163, BID, GABRB1, HDAC9, ARRB2, NPAS2, NDUFS1, ALDH1A1, ST8SIA4, PLA2G7, IMPA2, IGF2, NNMT, AMACR, NQO2, NGFR, ESS2, IL2RA, PRDX6, TRRAP, RASD2, ALOX12, DYM, LRP8, SLC30A3, MYH9, TSNAX-DISC1, NTSR1, SEMA3A, SAT1, FABP5, BCL2, FABP3, HRH2, PTPRA, FAAH, SNAP29, HSPA9, SYT11, ATM, NPY1R, TREM1, HTC2, ATF4, HTR1D, ZNF74, DBI, NEUROG1, GSTA1, TACR1, CKB, GNB3, LSAMP, CHRNB2, ACP1, PNOC, PITX3, ADARB1, MDH1, HCAR3, SYN1, CHRM2, GCH1, CDC42SE2, PRKCA, CHGA, GRB2, PLAT, STX1A, ADNP2, LILRB1, IL18R1, CRH, LPL, LYRM4, ACSM1, NALCN, GCLC, LMX1B, ENO2, SLC25A1, PLG, LMX1A, HTR3E, RGS2, LRP2, GFRA2, CYP3A5, SLC6A5, GC, GRM4, TACR3, MICB, TDO2, MIF, NPTN, REST, GABRA6, BLOC1S3, SLC7A10, LASP1, HPS4, CSNK1E, NPAS4, GSK3A, PEMT, MBD2, GSN, DICER1, TOP3B, CTNND2, ARHGAP18, CSMD2, FOLH1, TAP2, PRODH2, CACNG8, PANX2, STH, CACNG5, SMAD5, GRIA3, SLC6A13, PNPO, NDUFV1, FADS2, H2BC11, CABIN1, HOMER2, NDP, PER3, NRXN2, CDK5R1, MBNL1, MPZL1, PEX19, VPS39, SOX6, RANBP1, ST6GALNAC1, RGS5, ATP6, RBP1, CD99, ITGA8, MCTP2, GJD2, GNPAT, MAGI1, RELA, MOBP, GRIP1, GABRQ, TRPM1, TMSB10, ME2, PI4K2B, TNFSF13, MYO5B, PARL, MC5R, TIGAR, PLSCR4, MMP3, NUMBL, WDR11, RNR2, MTR, GPR50, LARS2, SIRT5, DAAM2, PAG1, GABARAP, AKAP5, VAMP1, VAMP7, DROSHA, FBXL21P, TAC1, B3GNT2, TAP1, TAPBP, CSPG5, SERPINI1, TCP1, IGF2BP2, AUTS2, GAS2L1, CDC42EP3, MAPK8, TFAP2A, TGFBR2, SLC6A11, PHB, THBS1, AKR1A1, VAMP2, MCAT, BRAP, STXBP1, TRMT2A, PPIA, PPARG, CNPPD1, SLIT3, SNCB, DKK4, PMP2, DKK3, PADI2, KLK8, GLS2, FSTL1, PTPN21, SPTBN2, SRD5A2, FXYD1, IL17C, GPR78, SRSF10, AP3M1, DNMT3L, IL19, PSAT1, TIMP4, ATXN2, PTPN6, GHRL, WNT2, RB1CC1, RIMS3, XRCC4, RAPGEF5, SLC38A2, ARHGAP32, RIMS2, S100A9, NPAS1, DGCR8, MAGEL2, DLL4, SH3PXD2A, ADAM12, CELSR1, ENTPD4, PADI4, MSRB1, NFASC, DCDC2, TPI1, PDZK1, TLR2, GEMIN4, PLEKHA6, TSPAN8, CALY, SHOX2, SHMT1, SBNO2, TPT1, SV2A, HNRNPA3P1, PCK1, PCDH8, SDHA, PTGDS, MYO16, PTGER3, OXSR1, CLDN11, SETDB1, HERC2, FRA7G, CCND2, PANK2, CD14, IDE, GRK6, CD48, GPS1, SEPTIN7, NDUFS7, EN2, CDK5, CDKN1C, NAV1, CYP26C1, GRB10, GRIK5, CCDC68, HRH1, LINC02694, NAV2, C1QB, EFCAB11, RETREG2, R3HDML, HAPLN3, DGKH, C4BPB, MIR326, IL2RG, CACNA1F, MIR185, CNIH3, PALB2, MIR130B, MIR107, CASP3, PDIK1L, CAV1, CBS, CHEK1, EN1, LMAN2L, TXNDC5, CGNL1, SEMA3D, DBN1, KREMEN1, EIF2S1, HNRNPA3, DIO3, DLX1, FBXO45, H1-4, H1-5, H2AX, DUSP6, SNX31, KAT8, SLC30A8, HAGH, HSPA12A, CTSK, CTRL, DIXDC1, HCAR2, HMGA1, CHRM5, ADAMTS12, HLA-J, EMX2, YWHAZP5, GSS, OFCC1, CREBBP, CREM, PLA2G4D, CRMP1, IFNL3, GSTT2, CSNK1D, MIR382, C4BPA, C1QA, BTBD9, ELOVL5, FLNB, PKNOX2, APBA2, APC, C1QTNF3, FGF2, FCGR2A, MIR2682, APOH, FAS, ARHGAP1, MIR497, KIF2A, DLG2-AS1, FBP1, FN1, GABRR1, KCNQ3, ADK, ABCA1, ACHE, FUT8, MSS51, LRP1, GABRA5, FADS1, ADORA1, LCN2, LIFR, GRK3, ADSS2, JAG1, ALDH3A1, ALDH3B1, ALK, KDR, ASAH1, ACSL4, WNK4, ERVK-8, BRCA1, BRCA2, BSG, BTC, CLVS2, WNK3, BIK, B3GAT2, GLRA2, KLF5, BTG1, CCDC86, IL12B, AGXT2, ITGB3, SNORD103C, USP46, KCNJ10, GFRA1, KCNJ3, KCNH1, KCNB1, GFRA3, B2M, PLA2G4B, MCHR2, CADPS2, JARID2, JAG2, CBLIF, TEKT5, OR4C46, SLC26A8, FAM3D, KDM2B, SAP30BP, LPAR1, GABRA4, GRIN1, BLOC1S1, ZNF480, SLC26A7, GABRD, EIF5, PML, KCTD12, ESAM, SLC23A3, RGS12, ZNF565, A1BG, MIR328, EDEM2, RNANC, MIR219A2, LMOD2, CDK16, DFNB47, INPP5A, SBNO1, MAGEC1, ITGAM, ACOT6, ND4, DEL22Q11.2, SEPTIN11, UGT1A3, KPNA1, PITPNM1, NRIP1, APOA4, LAMA2, FASTKD5, ERVW-4, ADAMTS3, BHLHE40, UNC5C, ADCY7, SOCS2, CASP4, CHD4, CCNA2, CP, SPATA5, TRAK1, ST3GAL1, HTR3D, PLLP, ADGRF4, SDF4, GPR153, ALS2CL, SNAP25, LAMA1, HLA-E, CFAP65, COL3A1, DAZAP1, NPRL2, CELF2, CETN1, ZNF530, CCDC137, KCNH6, YWHAZ, HIVEP2, GOT2, PPP3CB, NCSTN, CHST3, CTSC, PPM1G, NGF, ATP5F1C, KDM5C, KDM1A, ZNF521, ACTR3, ASH2L, SLCO6A1, SLC39A8, USH1G, KALRN, NT5C2, ATXN1, AS3MT, RBFOX1, MECP2, ITIH4, RENBP, SATB2, QPCT, CALB2, CALN1, ZNF365, EHMT1, SYNE1, MRTFA, CTNNA3, SNCA, AKT3, MMP16, BTN3A2, EFNA5, GABBR2, TMTC1, FURIN, HLA-DQA1, RAP1A, CACNA1I, SNAP91, DGKZ, ARHGAP4, TRANK1, ZFPM2, TAOK2, SF3B1, PRRC2A, RARG, KIT, CADM3, CLCN3, PDXDC1, ZBED4, SEC24C, PIK3C2A, FXR1, MEF2C, KLC1, PCDH15, UPF3B, RTN1, HLA-DRB5, ANKS1B, MSH5-SAPCD1, SLC4A10, BCL11B, RBM12, FOXO6, CUL3, FOXO3, TCF20, PRKN, SPATS2L, STT3A, HLA-DOB, NLGN4X, NEGR1, FAT3, ST3GAL3, FOXP1, ADCK1, DECR1, ATF7IP, YWHAG, CIB2, DLC1, MSI2, HLA-DMA, ZEB2, LRP1B, SPHKAP, NOSIP, NFATC3, PITPNM2, DNMT3A, CACNA1D, ZSWIM6, CAMK1D, ARSA, TBC1D5, CADM3-AS1, ZNF823, CCDC192, SHISA9, CTIF, FER1L6, MIR29B2CHG, GALNT10, SLC8A1-AS1, MROH6, TRAF3IP2-AS1, LINC00461, MAML3, ZSCAN16-AS1, CARMIL1, CNNM3, SMPD3, NDRG4, CCDC88C, FLOT1, DENND6B, GTDC1, TET2, RRN3, GRTP1, CARF, MTCO3P1, LRRN3, KCNMB2, DENND4A, STAG1, VWC2L, LINC00243, LINC02232, LINC00898, CCDC102B, PDZD7, ATAT1, BTN2A2, LINC01239, MPHOSPH9, RBM6, GGNBP1, GID4, CENPM, ARHGEF10L, MSTO1, DMTF1, RBM23, SPATA6L, ACTG1P22, GPC6, LILRP2, FAM184A, USH1C, TTC12, NSD3, MAIP1, TRIM10, GIPC2, PDIA6, CORO7, FARP1, SLC17A4, LINC02551, LINC00240, LINC01884, LINC02326, DGKI, LINC02210-CRHR1, LINC02822, CACHD1, LINC01905, RERE-AS1, LINC02188, LRRFIP1, CWC22, SMIM15-AS1, C2-AS1, LINC01583, LINC02109, CXXC4-AS1, MEF2C-AS1, LINC01876, TSBP1-AS1, LINC01149, ADIPOQ, NEURL1, SIPA1L2, LINC02360, DNAJA3, H2BP5, GRM3-AS1, TENM2, LINC02055, LINC02730, RN7SL100P, LINC02057, LINC02797, GRAMD1B, LINC02511, LINC02264, HIP1R, LINC02267, LINC02033, TTLL11-IT1, SLC7A6, LINC02571, HECW2, LINC02829, LIPC-AS1, KCNIP1-OT1, TENM3, DCAF1, CORO7-PAM16, ARHGAP15, BORCS7-ASMT, MUC22, RALGPS1, PLCH2, PRORP, LIN28B, PROX1-AS1, LINC01592, HDAC2-AS2, STARD4-AS1, CPEB1, LINC01539, ENPP7P7, WDR12, LINC01004, ZNF536, CLSTN3, GIGYF1, RGS6, HSPE1-MOB4, RBKS, CKLF-CMTM1, SCAF1, TGFBRAP1, HS6ST1, MED26, LINC01877, BBX, LINC01360, LINC02466, AKAP6, EP300-AS1, LINC01470, PAK6, STXBP5L, MRPL33, LINC00457, MPP4, EXOC4, PRDM14, CDH23, MIR100HG, UXS1, SFTA2, ZNF385B, IGSF11, SMG6, CNTN4, DNAJC13, ZNRD1, KCNIP1, PPP1R13B, MAU2, AGBL4, KLHL23, SLC9B1, ZNF664, LINGO1, C11orf21, CDH20, ATP6V1E2, ZNF638, PCED1B, DSG4, C12orf65, LSM1, ABCA5, CHST11, AMOTL1, FER1L6-AS2, TTLL11, IGSF9B, CNOT1, GLIS3, NTM, SLC7A6OS, DOP1A, SAMD4A, ASAP1, COA8, SMG1, OTUD7A, KDM4C, TBC1D2B, DDN, CCDC122, ZCCHC14, SLC45A1, MAML2, MYO18B, CCDC60, CYHR1, LINC00637, RFLNA, LRRN2, TMEM132D, ZBED9, MTHFD1L, TMEM219, OSBPL10, HYKK, DNAH1, SNED1, NRAD1, ANO4, LTN1, PPP1R16B, CABLES1, B9D1, LAMP3, C10orf90, RAB11FIP5, GIGYF2, ARHGEF26, EPC2, PHACTR3, ZBTB20, IP6K3, WHRN, MOB4, TRAPPC3, BACE2, TTC33, TRPM6, COX11P1, LETM2, MUCL3, RFT1, NDUFAF2, SNX29, PLA2G15, C3orf49, SPECC1, POM121L2, MTERF4, ZFAND2B, RBM45, CNTNAP5, SFXN5, PRPF40B, CEP41, OSCP1, NGEF, PCNX1, PSORS1C1, SORCS3, XKR6, CCDC39, KDM3B, EEPD1, MROH2A, TMPRSS5, OR5V1, SUFU, TAAR6, RFESD, NSMCE2, FAM86B3P, EFR3B, LINC00606, SLC17A3, IQGAP2, MPP6, C3orf38, CYB561D1, PDE10A, LINC00303, HCP5, C1orf167, ZKSCAN3, COQ10B, MSANTD1, EYS, SEMA6D, IPO8, SNORC, UBE2Q2P1, SPTLC1, TEX36, LRRTM4, ERLIN1, H1-12P, CCDC170, ZFAND6, GUSBP2, THOC7, SNTG1, HCN1, AGAP1, FTCDNL1, IFT74, SPG11, TSBP1, SOX2-OT, ACTL7A, OR12D3, EHMT2, PCDH12, ZDHHC2, CKLF, CADM2, ZSCAN23, HS3ST5, LEMD2, ZDHHC9, CCDC162P, NSUN6, R3HDM2, JMJD1C, TMX2, ADGRV1, SPRED2, ANKRD23, ADAMTS16, ARSG, PSORS1C2, EPN2, ASCC1, CEP78, FAM178B, TWF2, RAPGEF4, IMMP2L, TRIM31, ZNF615, SEH1L, ZNF93, DNAJB4, TTLL6, BTN3A1, BTN2A1, LINC00922, KATNAL2, FAM177A1, KSR2, RASSF1, MYO1H, ZNF740, JAKMIP3, SELENOH, SFMBT1, COMMD10, VPS45, FAM53C, DENND5B, H3P40, HIRA, BRD3, TUSC3, CACNA1S, MAN2A1, HLA-DRB2, ARL3, TEAD1, PSAP, KRT81, KRT83, ARSD, TEP1, KRT86, ALMS1, CYP2D7, ZSCAN9, PSMB9, TAF15, PRRG2, TBXAS1, PEPD, PGC, H2BC15, H2AC6, KCNN2, H2AC10P, PTPRF, STAU1, PTGIS, HLA-DRB9, HLA-DRB6, HLA-DRB3, MAP4, CNTN2, APBA1, TRIM26, NEK1, SRPK2, CLRN1, NEU1, BNIP3L, NFKB1, NFIB, NKX2-1, BMPR1B, LRP4, NFATC2, LMO7, LIPC, HARS1, FLI1, PRKAA1, GSDME, SCN4A, USH2A, DNTT, PRKG1, TSPO, PRKD1, HLA-DQA2, DRD5, HLA-DMB, GULOP, AFF3, HK2, C2, PLCB4, DPYSL2, PLCL1, TH, CYP21A2, RERE, ATP2B2, PTPRN2, DBH, HAT1, EIF3F, PBX2, IL1RN, INHBA, BCAR3, NOL4, CMAHP, MYO7A, CD40, ERCC8, SPG7, SGCD, TRIM27, NAB2, MYH11, STX2, STK24, ADCY1, RPN2, PLPP1, TRNE, ADAM10, RPL37, RGS4, RORA, SLC17A1, GALNT2, MGAT1, ATXN7, NPC1, ARHGEF7, OPRD1, GP1BB, EIF4G1, MSH5, STK19, FOXN3, EPHA5, ELAVL4, FES, ETF1, GBA, CDH13, CHRNA2, RREB1, EMX1, GNG7, AP2B1, PCDH7, TRNS2, PDLIM7, CFP, SGCG, GSTK1, TBC1D9, PIK3CD, MFGE8, POMC, PIK3CG, HMBS, BDNF-AS, CXCL8, PLB1, TSHZ1, ACKR3, CCL11, SSTR4, PLA2G2A, IGF1, CALM2, PGPEP1, BMP1, PRCP, EDNRA, ELK3, GPR42, HLA-DQB2, CALM3, BRS3, CXCR6, APP, ECT, SHANK2, ADRA2B, CAT, MARK1, CLINT1, LPAR2, HSPA4, SNRNP70, KAT5, DHDDS, RTN3, KRIT1, RARB, CDK9, SAFB, MIR132, CHPT1, FKBP5, EBPL, EGFR, ACACA, CAMKMT, BMS1, APOL1, SERPINA3, HTR1B, SCN2A, PTPN4, ATP5F1A, PRS, NOS3, KLF6, ACTG1, PSMG1, OR2AG1, PARP9, MIR34A, DNASE1L3, DUSP2, EP300, IL17A, DMD, F2R, DGCR, RAC1, NEK4, NPL, SYNM, ACTG2, SNX19, MRGPRX3, FZD4, SULT1E1, MACF1, PAK3, FXYD6-FXYD2, CIT, CUX1, EPHB1, MRGPRX1, PITX2, MIR183, INSRR, COX8A, SMS, ALDH2, LPAR3, GLP1R, MRGPRX4, GPR88, MIR195, ACOT7, CRYGD, EPO, MFAP1, PNPLA2, MAP3K8, MZB1, FHL5, APRT, GHR, DAXX, LGR6, P2RX7, KIDINS220, SERPING1, THOP1, STIN2-VNTR, POTEM, HCRT, COX1, SLC12A6, OXER1, GPHN, GPRC6A, LINC00273, ACTBL2, GPR151, FOS, GOLGA6A, PPT1, LEPR, WAS, LOC107987479, POTEKP, GAPDH, CYP2B6, CYP2C19, LINC01672, CXCR4, TERC, DRP2, VN1R17P, GPR166P, EPHB2, CNBP, SCZD13, PPARGC1B, NANOS1, TARDBP, RASGRF1, TERF2IP, GPR85, NPTX2, MCF2L, TREM2, NUP98, NTF4, PEBP1, RXRB, COL2A1, MTRR, RSRC1, RSS, RIT2, TLR9, FABP4, DHX9, ANGPTL2, ATN1, NEFM, DLL1, PRSS3, CALHM1, KCTD13, PRNP, IL27, DESI1, RABGEF1, CNTN6, DAB1, PRKCD, ARID2, NHS, PMP22, BLOC1S6, SEPTIN4, NF2, POU3F2, GNL3, DES, CX3CR1, DTNB, SERPINE1, PSMD9, CHDH, FBN1, GRIN3B, PDE2A, PDGFB, OR10A4, IL22, SERPINF1, PPM1K, A1CF, BORCS7, PYCARD, SDF2L1, PTK7, CRYM, SLC35G1, CSNK2B, CSPG4, TPSG1, ASPM, REM1, SERPINA1, OGG1, MSMB, H2AZ1, SEMA6A, LPP, PCDH19, MARCKS, AMBP, C3, CLEC10A, C4A, C4B, ALDOC, ALB, ITGA2B, ERMN, ARFGEF1, ABCG2, CA2, MIAT, ARID3B, ISG20, MDD1, AICDA, GNA13, AGT, MAPT, ASIP, A2M, MAS1, MIR223, ADRB2, HNMT, LIF, STATH, LGALS3, ROPN1L, OCLN, L1CAM, LAMC2, NR4A3, HAPLN2, APOA1, SLC14A2, VWF, SPRY4, VIM, ALDH7A1, UQCRC1, GDF15, HCL2, UCHL1, ADAMTS10, MGS, DLGAP3, UBE2N, PLA2G12A, ABCB6, TYMS, TXN, OPN1MW3, ATG5, CCL4, OPN1MW2, ANGPT2, MIR219A1, PTGES3, STAT1, DLK1, C21orf62, HSP90AA1, CHP1, GLUD1, TMEM108, SMARCA1, MCPH1, SYNPO, IL2RB, GIPR, CD28, SH3GL2, HSPG2, GAS7, FAN1, GH1, GLT8D1, MALAT1, OPN1MW, CDK4, LOC110806262, SKAP2, SCZD2, CECR, IGFALS, CCL17, HSPA8, CD9, MEF2A, SPTAN1, SLC9A3R1, ARHGAP11B, UTS2R, LYVE1, KL, FTO, MDM2, WDHD1, CAMK2A, IL33, ASPA, GPR52, SOS1, CCKBR, PKP4, CHST9, ZBP1, F10, F5, ICAM3, NIPA1, DEGS2, IFNB1, HLA-G, HSPD1, HNRNPD, HNRNPC, HNRNPU, IFNGR2, FCN2, NETO1, GLYCTK, DNAJC5, HPD, SRCIN1, SLC38A1, DNAJB1, FGF9, PTK2B, HSD11B2, FGF10, FGA, HPS1, FGF14, HNRNPK, NR4A1, FARSA, FAT1, IARS1, HSPA5, H3-3A, HLA-DRA, GLUD2, GABPA, G6PD, PANX3, FUCA2, GMFB, GNAQ, ISX, FRAXA, PKDCC, APOA5, FRA6F, AGAP3, GPC1, GPR17, GPT, CHURC1, ARHGAP33, TMPRSS13, GABRG1, CDCA5, TXNRD3, IL17F, MYL12B, GBP1, GCHFR, SLITRK1, GART, GCSH, GGTA1P, GALC, MARCHF9, ASB16, GHRH, GLDC, LMLN, CXCL1, FXN, TMC2, HSD17B10, FLT1, HAL, HCFC1, FOXG1, SARNP, ATP13A4, HTT, CFH, FKBP4, H4-16, FH, HIF1A, UBE2K, FGFR3, HABP2, H3-3B, COMTD1, GUK1, GRP, GSPT1, GTF3A, SLC22A16, EXOSC6, TRIM5, ATCAY, LMNB2, FOSB, UBASH3B, GUSB, FBH1, GYPC, PHF5A, GNB1, CRYBB2, EZH2, BLVRB, CFB, SNORD118, POTEF, OPN1SW, MEIKIN, OIP5-AS1, CCR2, TNFRSF17, PSS, DDTL, TLX1NB, MIR936, MIR708, BCHE, BBS4, BAGE, TSTD1, ATP6V1A, ATP2B4, DIP, AUTS1, ATF3, SERPINC1, WASHC1, ZNF729, C4B_2, MIR1288, IQCJ, GSTT2B, MIR1271, WASH6P, SLC25A20, MIR22, MIR25, MIR301A, MIR30A, MIR30B, CACNB4, MIR320A, MIR9-2, MIR98, CACNA1A, EIF2AK4, CDNF, DAGLA, VPS51, C5, MIR338, PLF, H4C15, MIR485, BTN1A1, MBOAT4, DST, POU5F1P3, BMP6, H3P16, POU5F1P4, MIR1307, MIR664A, EZH1, LOC102723407, ADRA2C, CBSL, ADRA1D, PARP1, LOC102724971, TPTEP2-CSNK1E, ADH5, ADD2, THRA1/BTR, ADA, UPK3B, CERNA3, ACTC1, ACP3, ACO2, ACADS, FMR1-IT1, ABO, MTCO2P12, AOC1, ABCF1, DM1-AS, ERVK-32, NAT2, H3P8, H3P24, H3P19, LOC102724334, AFM, ASTN1, ACAN, FAS-AS1, MIR3162, ASCL1, STS, ARNT, ARF1, AQP3, P2RX5-TAX1BP3, MIR4449, ERVK-20, ANXA6, ANXA3, SPRY4-IT1, ANXA1, SLC25A4, IFNG-AS1, COMETT, NOTCH2NLC, NOTCH2NLB, AMY1A, AMELX, ALCAM, ALAS1, AKT2, AK1, AGTR1, AGRP, MIR212, MIR204, MIR200C, ARX, DOCK1, DNASE1L1, DNASE1, DNAH9, GABRR3, DLD, DIAPH2, TUBB, DHFR, DCX, OPN5, DCR, BRINP1, DAP, CYP19A1, YTHDF3, CYP2C9, LRRC57, CST3, CSNK2A1, CRYBB1, NEAT1, KCTD21, TTC9C, CRY2, CRY1, CRK, FUT11, DPYS, MIR19B1, DR1, EXT1, UBXN2B, ETFA, ESRRA, BPIFA2, NRSN1, ESD, ISM1, SIRPA, ERG, PIWIL4, ERCC5, ERBB2, PWAR1, NRG4, EPHB4, LYPD4, EPB41L1, ELANE, EIF4A2, EIF2S3, KCTD7, EIF2D, CLVS1, S1PR1, EDA, MARCHF10, CRIP2, CRIP1, CRHR2, PGP, CD36, CCL4L1, MS4A1, NOTCH2NLA, CD19, CD8B, MUC21, CD4, CCT6A, CCNH, KYAT1, RUNX1, HAPLN4, LINC01194, PHOBS, MIRLET7G, CASP1, MIR125B1, MIR126, CAMP, MIR144, MIR146A, MIR15B, CALR, MIR184, MIR193A, MIR198, CD38, CD59, CD68, CHRNA1, CRHBP, ENDOV, CPS1, CXCL17, CPOX, CORT, MIF-AS1, COL9A3, SUMF1, CLTB, TBCB, TMED10P1, CLEC4D, CD79A, CHRM3, LYST, CGA, NAT8L, CFL1, ATP13A5, CDKN2A, CDKN1A, TUBB2B, CDH15, CLAM, CDH11, SYNDIG1, ENDOU, CNTNAP3, STAR, SRPRA, WDR3, SRY, SLC26A1, SSTR5, PGRMC1, ALDH1L1, TBR1, STRN, ZMYND11, KDM5B, NES, STXBP3, SULT2A1, UTS2, SUB1, EHD1, HNRNPA0, TMED2, MLLT11, RAB40B, TMED10, METAP2, IMMT, SLC38A3, SLC27A5, SRM, SRF, STMN2, TPPP, SPR, ADAM17, EBP, SPN, THAS, NXF1, ATP5PD, BAIAP2, VAV3, TGFBI, IFITM3, THBD, TAL1, TUBA1B, THRA, THRB, TIMP1, KATNB1, MARCHF6, UNC13B, SEMA4B, TGFB2, TFPI, ARL6IP5, TERT, SORBS1, TXNRD2, AHSA1, TRIM3, TXNIP, PRDX2, TCTA, TRGVB, ZEB1, HNF1B, TBCD, RPP14, SPARC, TIMP3, FBXO28, RPS10, WWC1, POFUT2, ANKRD12, RPS19, SORT1, TBC1D1, SCD, S100A10, S100A11, STAB1, MAPK8IP3, SEPTIN6, EPB41L3, RPS6KA3, CUX2, RPL13, RPL3, RPA1, RORC, RNH1, RLBP1, RFX1, REN, REG1A, RBP4, ABCB9, BRD4, RANGAP1, POFUT1, MOK, ATXN8OS, NCOA6, CD160, SMN1, SGK1, SHBG, SKIL, SKP1, SLC2A3, SLC16A1, COPE, CCL21, SMN2, HRH3, SNRPN, NISCH, SORL1, CAPN10, MMRN1, SEPHS1, RAB3GAP1, DKK1, P2RX2, TRIM32, SFSWAP, SRSF7, PDCD11, SRSF6, SET, SELENOP, GOLGA8A, SELL, CXCL12, SCZD1, CCL22, TIMP2, STUB1, PVR, H4C13, H4C3, H4C8, ZRANB2, GRAP2, H4C2, H4C5, COX5A, TGM5, LOH19CR1, KIF3B, PLA2G10, ULK1, LHX2, STX7, H4C11, GSTO1, H4C12, H4C6, ARHGEF6, H4C4, H4C1, H3C6, CABP1, CHST10, TBX4, ADAMTS4, ADAMTS2, FZD7, TBPL1, GOSR1, NRG2, TCEAL1, BBOX1, FZD1, SLC25A14, CLDN12, MAP7, CCRL2, CES2, TMEM11, CCN6, DLEU2, TRIP4, KAT2B, NR1I2, WASF1, FUBP1, BSN, SNURF, RIPK2, CLDN1, ATP6V0D1, INA, P2RX6, KCNK5, USO1, ARHGEF2, BCAS1, ZMYM3, XPR1, VAPB, CDC42BPA, NR0B2, PIWIL1, KLF4, TRIP13, CCL4L2, PICALM, CDK2AP2, SUMO1, TTN, TYRP1, UBE2D1, SRA1, NAALAD2, UBE3A, HNRNPDL, TRPV1, THOC1, NR1H4, UCP1, WDR1, ARNT2, VCAM1, TSC2, BCAP31, TRIO, AKAP9, EBI3, CCT3, TRC-GCA24-1, WASF2, TPP2, ALYREF, TPO, TOP1, TLR5, TLR1, RIDA, HCG9, TKT, BEST1, VRK1, H4C9, VGLL4, IL1R2, GPRIN2, ULK2, FZD5, RAPGEF2, DEK, IQCB1, KIAA0319, EPM2A, AIMP2, SLC7A5, ABCG1, MIA, USP9X, DNALI1, ZNF45, ZSCAN12, FRMPD4, YWHAB, TOX, KIAA0513, PCLAF, YY1, XRCC3, XPC, RNF40, WNT7A, SFI1, WNT3, WNT1, DNAJC6, SEC14L2, PURA, NANOG, CTNNBL1, RETN, MLLT3, KCNQ5, EIF4ENIF1, NR3C2, KYAT3, BTNL2, MNAT1, PCDHA1, ALG1, MYDGF, CFC1, NSFL1C, LANCL2, FAM20C, EIF2S2, ADAM11, CD46, MCM7, MCC, MC4R, MC2R, MAN1C1, VARS2, MAP1B, MAP1A, AMIGO1, SMAD4, SMAD3, ESYT2, HEG1, GPRC5C, USE1, LRFN2, GOLPH3L, GPRC5D, MYO1D, MEG3, TMEM176A, CCDC25, NDUFB5, NDUFS3, MPI, NDUFS6, NDUFV2P1, NEDD4, SEPTIN2, WDR60, MARCHF1, MYD88, MUC1, KIF21A, PPP1R9A, MTTP, IPO9, ND5, ND1, COX2, MT1B, MST1, MBD5, MS, CACNA2D3, LRP2BP, MPZ, MPP2, MRTFB, CYP4F3, NCAPG2, ISL1, IL15RA, NUP37, GGCT, INSR, UBE2Z, IRF3, ITGB1, KCNQ1, NT5DC2, ITGB4, ITGB5, ANOS1, PDIA2, NUCKS1, IL12A, PLEKHF1, IL9R, EFHD2, MMEL1, IL9, SLC52A2, IL6ST, SNIP1, ZNF750, IL1R1, ALG9, RBPJ, IGHG3, IGHG1, IGFBP4, HDAC11, GINGF2, KIF5A, LNPEP, LCT, LBP, ACE2, IL21, LCK, LCN1, KMT2C, CXCL16, KIR3DL2, GRHL3, POLD4, SUGP1, LGALS1, CHD8, LIG4, CACNG6, HRH4, SCPEP1, STMN1, RPSA, GAS5, BCAN, BLOC1S5, SCZD10, KIF13A, NEUROG2, KTN1, MCCC2, LRRC4, VSIR, KRT7, KNG1, NEFH, WBP1L, CDC42EP4, SEPTIN5, PLXNB3, PMCH, AGO2, NDOR1, CYTH4, PMM2, EIF2AK1, PTPA, CACNG4, POLR2A, POU3F1, POU5F1, NSG1, LAT, PLXNB1, SERPINF2, PLEC, GNMT, PLCG1, APEX2, PLA2G5, TMEM97, SGSM3, PCLO, PKHD1, TRBV7-2, TRBV2, IGLV3-25, LAMTOR2, GIT1, SETD2, PPP2CA, PPP3CA, SERPINA4, PTEN, PYY, PART1, PSMD10, PSPH, SPDEF, TAS2R38, TBC1D22A, CYFIP2, PANX1, PTPRG, CD2AP, PTPRZ2, PDSS1, SMUG1, MXRA5, RNF19A, PSMB6, ATL3, CNRIP1, MOXD1, PROS1, PSPN, MAP2K1, MAPK4, POLDIP2, PRKCG, PRKAR2B, PRKAR1A, FGF21, PREP, ELP4, TAGLN3, PADI1, NEUROD1, NRCAM, NPTX1, H2BS1, CNTN5, NPY2R, NRAS, IL17D, NUCB2, OPRL1, NVL, TNFRSF11B, TPPP3, SF3B6, SRRT, IL23A, NPPA, NLGN3, ANLN, KRT20, NOVA1, CCHCR1, NOTCH3, NOTCH2, UGT1A8, MINDY2, NOS2, NONO, CRLS1, NMBR, NFE2L2, NEUROD2, GATAD2A, SCLY, P2RX1, PGK1, HP1BP3, ST8SIA3, PCCB, TMED5, SERPINA5, F11R, DHH, PCMT1, SUCO, PCYT1A, PDE9A, ITSN2, PENK, VSX1, PFKM, MECR, PAEP, ANGPTL4, INSIG2, HSD17B12, AADAT, P4HB, P2RY2, P2RY1, BFAR, TLR7, RASL12, P2RX5, P2RX4, HOOK1, PLA1A, P2RX3, H4C14

-
Spastic Paraplegia, Intellectual Disability, Nystagmus, And Obesity
Omim
Description Spastic paraplegia, intellectual disability, nystagmus, and obesity (SINO) is an autosomal dominant neurologic disorder characterized by rapid growth in infancy, global developmental delay, spastic paraplegia, variable ophthalmologic defects, and dysmorphic facial features (summary by Josifova et al., 2016). ... All had significant growth during the first year of life, resulting in height, weight, and head circumference in the high 90th percentiles. ... Molecular Genetics In 3 unrelated patients with SINO, Josifova et al. (2016) identified 3 different de novo heterozygous truncating mutations in the KIDINS220 gene. The first 2 mutations (W1350X, 615759.0002; Q1366X, 615759.0002) were found by next-generation sequencing analysis of a targeted gene panel, and the third mutation (c.4530dup; 615759.0003) by whole-exome sequencing. Analysis of cells from the first 2 patients showed that their mutations resulted in the production of truncated proteins that were similar to functional KIDINS220 splice variants with alternative terminal exon splicing as described by Schmieg et al. (2015). ... These changes also occurred in the last exon of the gene, suggesting that not every loss-of function variant causes the phenotype. INHERITANCE - Autosomal dominant GROWTH Height - Increased height compared to age-matched controls, postnatal Weight - Obesity, postnatal HEAD & NECK Head - Enlarged head circumference - Brachyplagiocephaly - Prominent forehead Face - Full cheeks Eyes - Nystagmus - Poor visual acuity - Hypermetropia - Astigmatism - Squint - Esotropia - Deep-set eyes MUSCLE, SOFT TISSUES - Axial hypotonia - Limb hypertonia NEUROLOGIC Central Nervous System - Delayed psychomotor development - Intellectual disability - Delayed speech - Spastic paraplegia - Hyperreflexia - Enlarged lateral ventricles - Cerebral atrophy - Reduced white matter volume - Delayed myelination - Partial agenesis of the corpus callosum (1 patient) MISCELLANEOUS - Onset in infancy - Three unrelated patients have been reported (last curated January, 2017) - De novo mutation MOLECULAR BASIS - Caused by mutation in the 220-kD kinase D-interacting substrate gene (KIDINS220, 615759.0001 ) ▲ Close
-
Railway Spine
Wikipedia
Railway spine was a nineteenth-century diagnosis for the post-traumatic symptoms of passengers involved in railroad accidents . The first full length medical study of the condition was John Eric Erichsen 's classic book, On Railway and Other Injuries of the Nervous System . [1] For this reason, railway spine is often known as Erichsen's disease . ... Germany's leading neurologist, Hermann Oppenheim , claimed that all railway spine symptoms were due to physical damage to the spine or brain, whereas French and British scholars, notably Jean-Martin Charcot and Herbert Page, insisted that some symptoms could be caused by hysteria (now known as conversion disorder ). [2] [3] [4] Erichsen observed that those most likely to be injured in a railway crash were those sitting with their backs to the acceleration. ... Institute of Railway Studies and Transport History . Retrieved 2008-08-27 . ^ Page, Herbert (1885). Injuries of the Spine and Spinal Column Without Apparent Mechanical Lesion . ... Churchill. pp. 25 , 62, 69, and 162. page Injuries of the Spine and Spinal Cord Without Apparent Mechanical Lesion. ^ Scaer, Robert (2007).

-
Cortical Dysplasia, Complex, With Other Brain Malformations 3
Omim
Molecular Genetics In 2 unrelated patients with complex cortical dysplasia and microcephaly, Poirier et al. (2013) identified different de novo heterozygous mutations in the KIF2A gene (H321D, 602591.0001 and S317N, 602591.0002). The first mutation was found by whole-exome sequencing and was not present in several genomic databases, including dbSNP, 1000 Genomes, the Exome Variant Server, and a local Paris Descartes Bioinformatics platform database. ... Because KIF2A functions as a dimer, Poirier et al. (2013) postulated a dominant-negative effect. The findings extended the association between microtubule-based cellular processes and proper cortical development. INHERITANCE - Autosomal dominant GROWTH Other - Intrauterine growth retardation (1 patient) HEAD & NECK Head - Microcephaly Eyes - Nystagmus (1 patient) NEUROLOGIC Central Nervous System - Delayed psychomotor development - Seizures - Spastic tetraplegia - Malformations of cortical development - Thick cortex - Subcortical band heterotopia - Pachygyria - Agyria - Thin corpus callosum - Dysmorphic basal ganglia (1 patient) MISCELLANEOUS - Two unrelated patients have been reported (last curated September 2013) MOLECULAR BASIS - Caused by mutation in the kinesin heavy chain member 2A (KIF2A, 602591.0001 ) ▲ Close





