Summary Clinical characteristics. Trichorhinophalangeal syndrome (TRPS) comprises TRPS I (caused by a heterozygous pathogenic variant in TRPS1 ) and TRPS II (caused by contiguous gene deletion of TRPS1, RAD21, and EXT1 ). ... Clinical Characteristics Clinical Description Trichorhinophalangeal syndrome (TRPS) comprises TRPS I (caused by a heterozygous pathogenic variant in TRPS1 ) and TRPS II (caused by contiguous gene deletion of TRPS1 , RAD21 , and EXT1 ). ... Giedion suggested the name tricho-rhino-phalangeal syndrome because of the triad of most prominent features [Giedion 1966]. ... Hall et al [1974] suggested subdivision of TRPS into TRPS I for individuals with normal development and absent osteochondromas, and TRPS II or Langer-Giedion syndrome for those with intellectual disability and multiple osteochondromas. ... Disorders to Consider in the Differential Diagnosis of Trichorhinophalangeal Syndrome View in own window Disorder Gene(s) MOI Clinical Features of the Differential Disorder Overlapping w/TRPS Distinguishing from TRPS Oculo-dento-digital syndrome GJA1 AD Slow-growing, dry hair Underdeveloped alae nasi Long philtrum Eye signs Dental signs more expressed Cartilage-hair syndrome RMRP AR Short stature Fine hair Cone-shaped epiphyses Nasal shape Immunodeficiency Ellis-Van Creveld syndrome EVC EVC2 AR Short stature Brachydactyly Nasal shape Oral frenula Polydactyly AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance Management Evaluations Following Initial Diagnosis To establish the extent of disease and support needs of an individual diagnosed with trichorhinophalangeal syndrome (TRPS), the following should be evaluated.
A number sign (#) is used with this entry because Witkop syndrome is caused by heterozygous mutation in the MSX1 gene (142983) on chromosome 4p16. ... Wicomb et al. (2004) documented the manifestations of Witkop syndrome in an affected child and his father. The paternal grandfather was also affected. Inheritance Witkop syndrome is inherited as an autosomal dominant trait (Hudson and Witkop, 1975; Jumlongras et al., 2001). Mapping Jumlongras et al. (2001) found linkage between the tooth-and-nail syndrome, which they referred to as Witkop syndrome, and polymorphic markers in the region of the MSX1 locus (142983) in a 3-generation family. Molecular Genetics In a 3-generation family with Witkop syndrome, Jumlongras et al. (2001) identified a nonsense mutation in the MSX1 gene (142983.0003) that cosegregated with the phenotype.
Hypodontia-nail dysplasia syndrome is a form of ectodermal dysplasia. ... Sweat glands, heat tolerance and hair are normal, although fine hair has been described in some cases. Etiology The syndrome is caused by a mutation in the MSX1 gene (4p16.1). Diagnostic methods Diagnosis is usually made during mid-childhood when persistence of the primary dentition becomes apparent, but the syndrome may be difficult to diagnose as the nail defects may be mild.
Yemenite deaf-blind hypopigmentation syndrome is an exceedingly rare genetic disorder characterized by cutaneous pigmentation anomalies, ocular disorders and hearing loss. Epidemiology The syndrome was described in 1990 in two patients from the same Yemenite family. ... Their parents were unaffected and possibly consanguineous. Etiology The cause of this syndrome has not been determined. Genetic counseling The inheritance pattern appears to be autosomal recessive.
A number sign (#) is used with this entry because of evidence that hypotrichosis-lymphedema-telangiectasia syndrome (HLTS) is caused by homozygous mutation in the SOX18 gene (601618) on chromosome 20q13. ... Description Hypotrichosis-lymphedema-telangiectasia syndrome is an autosomal recessive disorder characterized by these 3 features, which begin at birth or in early childhood and are progressive (summary by Irrthum et al., 2003).
Hypotrichosis-lymphedema-telangiectasia syndrome (HLTS) is a rare condition that, as the name suggests, is associated with sparse hair (hypotrichosis), lymphedema , and telangiectasia , particularly on the palms of the hands.
Tietz syndrome is a rare condition characterized by hearing loss, fair skin, and light-colored hair. The hearing loss in affected individuals is caused by abnormalities of the inner ear (sensorineural hearing loss) and is present from birth. People with Tietz syndrome are born with white hair and very pale skin but their hair color often darkens over time; The colored part of the eye (the iris) is blue.
A number sign (#) is used with this entry because of evidence that Tietz albinism-deafness syndrome (TADS) is caused by heterozygous mutation in the MITF gene (156845) on chromosome 3p13. A highly overlapping disorder, Waardenburg syndrome type 2A (WS2A; 193510), is also caused by heterozygous mutation in the MITF gene. ... Amiel et al. (1998) presented detailed clinical findings on the affected mother and son reported by Tassabehji et al. (1995) and noted that, although they fulfilled diagnostic criteria for Waardenburg syndrome type II (see 193510), they more closely resembled the family reported by Tietz (1963). ... Smith et al. (2000) reascertained the family reported by Tietz (1963) and confirmed the existence of the syndrome through at least 4 generations. ... INHERITANCE - Autosomal dominant HEAD & NECK Ears - Hearing loss, sensorineural, bilateral profound congenital Eyes - White eyelashes - White eyebrows - Blue eyes - No heterochromia iridis - Hypopigmented fundi SKIN, NAILS, & HAIR Skin - Fair skin Hair - White eyelashes - White eyebrows - White-blonde hair MISCELLANEOUS - Allelic to Waardenburg syndrome, type IIA ( 193510 ) MOLECULAR BASIS - Caused by mutation in the microphthalmia-associated transcription factor gene (MITF, 156845.0006 ) ▲ Close
Tietz syndrome is a genetic hypopigmentation and deafness syndrome characterized by congenital profound bilateral sensorineural hearing loss and generalized albino-like hypopigmentation of skin, eyes and hair. Epidemiology Tietz syndrome has been reported in 7 families to date. ... Psychomotor development is normal. Etiology The syndrome is due to a missense mutation or in-frame deletion of one amino acid in the basic domain of the MITF (3p14-p13) gene, coding a basic helix-loop-helix (bHLH) leucine zipper transcription factor, regulating melanocyte development and the biosynthetic melanin pathway. However, these types of mutations give rise to variable phenotype, ranging from Tietz syndrome to Waardenburg syndrome type 2 (see this term), with possible interactions with modifier loci. Genetic counseling Tietz syndrome is an autosomal dominant syndrome.
Tietz syndrome is a disorder characterized by profound hearing loss from birth, fair skin, and light-colored hair. ... The changes to the retinal pigment epithelium are generally detectable only by an eye examination; it is unclear whether the changes affect vision. Frequency Tietz syndrome is a rare disorder; its exact prevalence is unknown. Only a few affected families have been described in the medical literature. Causes Tietz syndrome is caused by mutations in the MITF gene. ... MITF gene mutations that cause Tietz syndrome either delete or change a single protein building block (amino acid) in an area of the MITF protein known as the basic motif region. ... Decreased melanin production (hypopigmentation) accounts for the light skin and hair color and the retinal pigment epithelium changes that are characteristic of Tietz syndrome. Researchers suggest that Tietz syndrome may represent a severe form of a disorder called Waardenburg syndrome, which can also be caused by MITF gene mutations.
Overgrowth. Sotos syndrome is associated with overgrowth of prenatal onset. ... Joint laxity is reported in at least 20% of individuals with Sotos syndrome. Maternal preeclampsia occurs in about 15% of pregnancies of children with Sotos syndrome. ... Other features that do not overlap w/Sotos syndrome are most similar to Weaver syndrome. ... Given the overlap in findings between Sotos syndrome, Weaver syndrome, Cohen-Gibson syndrome, and SUZ12 -related overgrowth, in probands with features suggestive of Weaver syndrome, the recommended testing order is NSD1 , followed by EZH2 , followed by EED , followed by SUZ12 . 2. ... A Sotos syndrome-like phenotype has been associated with 4p duplications, mosaic 20p trisomy [Faivre et al 2000], and 22q13.3 deletion syndrome.
Diagnosis Opitz et al. (1998) discussed the differentiation of 2 overgrowth syndromes, Sotos syndrome and Weaver syndrome (277590), and the question of whether the similarities are sufficient to consider them 1 entity. ... In Sotos syndrome there is remarkably advanced dental maturation; this is rarely commented on in Weaver syndrome. In Weaver syndrome, there are more conspicuous contractures and a facial appearance that experts find convincingly different from that in Sotos syndrome. ... Differential Diagnosis Schaefer et al. (1997) concluded that neuroimaging findings of Sotos syndrome are distinct enough to allow differentiation of this syndrome from other mental retardation syndromes with macrocephaly. ... Beckwith-Wiedemann syndrome (BWS; 130650) is, like Sotos syndrome, an overgrowth syndrome.
A rare genetic overgrowth syndrome characterized by a typical facial appearance, overgrowth with macrocephaly and variable intellectual impairment. ... Cancer predisposition is debated: the increased risk, if any, appears to be low. Etiology Sotos syndrome is caused by mutations or microdeletions in the NSD1 gene (5q35) in more than 95% of cases. ... It can be confirmed by molecular genetic testing of the causative genes. Overlap with other overgrowth syndromes may complicate diagnosis. Differential diagnosis The differential diagnosis should include Malan syndrome, Simpson-Golabi-Behmal syndrome, Weaver syndrome, Tatton-Brown syndrome, Bannayan-Riley-Ruvalcaba syndrome, Fragile X syndrome and megalocephalic syndromes associated with mutations in the PI3K-AKT-mTOR pathway. ... Management and treatment There is no specific treatment for the syndrome. Management of Sotos syndrome requires a multidisciplinary approach. Prognosis Sotos syndrome shows a wide spectrum of intellectual impairment, with functioning from fully independent to fully dependent.
A number sign (#) is used with this entry because of evidence that Sotos syndrome-3 (SOTOS3) is caused by homozygous mutation in the APC2 gene (612034) on chromosome 19p13. ... For a discussion of genetic heterogeneity of Sotos syndrome, see SOTOS1 (117550). Clinical Features Almuriekhi et al. (2015) reported 2 sibs, born of consanguineous Egyptian parents, with intellectual disability (IQs of 60 and 56), a severe receptive and expressive language disorder, learning disabilities, and hyperactive behavior associated with relative macrocephaly, long face, and prominent chin and nose. ... The findings were reminiscent of Sotos syndrome, but genetic analysis excluded mutations in the NSD1 gene (606681). The patients were diagnosed with a Sotos-like syndrome. Inheritance The transmission pattern of SOTOS3 in the family reported by Almuriekhi et al. (2015) was consistent with autosomal recessive inheritance.
Sotos syndrome is a condition characterized mainly by distinctive facial features; overgrowth in childhood; and learning disabilities or delayed development. ... Other signs and symptoms may include intellectual disability; behavioral problems; problems with speech and language; and/or weak muscle tone (hypotonia). Sotos syndrome is usually caused by a mutation in the NSD1 gene and is inherited in an autosomal dominant manner.
Raymond-Céstan syndrome Other names upper dorsal pontine syndrome , Basillar artery runs down the middle(in above image) and blockage is cause of this condition Raymond-Céstan syndrome is caused by blockage of the long circumferential branches of the basilar artery . [1] It was described by Fulgence Raymond and Étienne Jacques Marie Raymond Céstan . [2] Along with other related syndromes such as Millard-Gubler syndrome , Foville's syndrome , and Weber's syndrome , the description was instrumental in establishing important principles in brain-stem localization. [3] Contents 1 Presentation 2 Diagnosis 3 Treatment 4 References 5 Further reading Presentation [ edit ] Ipsilateral ataxia and coarse intention tremor (damage to superior and middle cerebellar peduncle ) Ipsilateral paralysis of muscles of mastication and sensory loss in face (damage to sensory and motor nuclei and tracts of CN V ) Contralateral loss of sensory modalities in the body (damage to spinothalamic tract and medial lemniscus ) Contralateral hemiparesis of face and body (damage to corticospinal tract ) may occur with ventral extension of lesion Horizontal gaze palsy may occur (as in lower dorsal pontine syndrome) Diagnosis [ edit ] This section is empty. ... "The crossed paralyses. The original brain-stem syndromes of Millard-Gubler, Foville, Weber, and Raymond-Cestan". ... Cite journal requires |journal= ( help ) Further reading [ edit ] Kim, JS; Lee, JH; Im, JH; Lee, MC (Jun 1995). "Syndromes of pontine base infarction. A clinical-radiological correlation study". ... Krasnianski, M; Neudecker, S; Zierz, S (Aug 2004). "[Classical crossed pontine syndromes]". Fortschritte der Neurologie · Psychiatrie (in German). 72 (8): 460–8. doi : 10.1055/s-2004-818392 . PMID 15305240 . v t e Cerebrovascular diseases including stroke Ischaemic stroke Brain Anterior cerebral artery syndrome Middle cerebral artery syndrome Posterior cerebral artery syndrome Amaurosis fugax Moyamoya disease Dejerine–Roussy syndrome Watershed stroke Lacunar stroke Brain stem Brainstem stroke syndrome Medulla Medial medullary syndrome Lateral medullary syndrome Pons Medial pontine syndrome / Foville's Lateral pontine syndrome / Millard-Gubler Midbrain Weber's syndrome Benedikt syndrome Claude's syndrome Cerebellum Cerebellar stroke syndrome Extracranial arteries Carotid artery stenosis precerebral Anterior spinal artery syndrome Vertebrobasilar insufficiency Subclavian steal syndrome Classification Brain ischemia Cerebral infarction Classification Transient ischemic attack Total anterior circulation infarct Partial anterior circulation infarct Other CADASIL Binswanger's disease Transient global amnesia Haemorrhagic stroke Extra-axial Epidural Subdural Subarachnoid Cerebral/Intra-axial Intraventricular Brainstem Duret haemorrhages General Intracranial hemorrhage Aneurysm Intracranial aneurysm Charcot–Bouchard aneurysm Other Cerebral vasculitis Cerebral venous sinus thrombosis v t e Symptoms , signs and syndromes associated with lesions of the brain and brainstem Brainstem Medulla (CN 8, 9, 10, 12) Lateral medullary syndrome/Wallenberg PICA Medial medullary syndrome/Dejerine ASA Pons (CN 5, 6, 7, 8) Upper dorsal pontine syndrome/Raymond-Céstan syndrome Lateral pontine syndrome ( AICA ) (lateral) Medial pontine syndrome / Millard–Gubler syndrome / Foville's syndrome ( basilar ) Locked-in syndrome Internuclear ophthalmoplegia One and a half syndrome Midbrain (CN 3, 4) Weber's syndrome ventral peduncle, PCA Benedikt syndrome ventral tegmentum, PCA Parinaud's syndrome dorsal, tumor Claude's syndrome Other Alternating hemiplegia Cerebellum Latearl Dysmetria Dysdiadochokinesia Intention tremor ) Medial Cerebellar ataxia Basal ganglia Chorea Dystonia Parkinson's disease Cortex ACA syndrome MCA syndrome PCA syndrome Frontal lobe Expressive aphasia Abulia Parietal lobe Receptive aphasia Hemispatial neglect Gerstmann syndrome Astereognosis Occipital lobe Bálint's syndrome Cortical blindness Pure alexia Temporal lobe Cortical deafness Prosopagnosia Thalamus Thalamic syndrome Other Upper motor neuron lesion Aphasia
Dubowitz syndrome is a very rare genetic and developmental disorder with a broad range of signs and symptoms. The typical findings of Dubowitz syndrome include growth failure/short stature, characteristic facial features such as a small triangular face, high sloping forehead, drooping eyelid (ptosis), short eyelids, increased distance between eyes (hypertelorism) broad and flat nasal bridge with a prominent and rounded nasal tip, smaller than normal head (microcephaly), intellectual disability, and eczema , especially on the face and behind the knees. ... The cause is still unknown, but, some people who are diagnosed with the syndrome may have variants (mutations) in the NSUN2 and LIG4 genes, or have loss or gain of microscopic material in some chromosomes ( chromosomal microdeletions or microduplications).
The case reported by Kondo et al. (1987) demonstrates that the syndrome occurs in Orientals as well as in Caucasians. ... The lack of a basic biochemical characterization of Dubowitz syndrome makes it impossible to be completely certain of the diagnosis in these cases. Lyonnet et al. (1992) commented on the paucity of information on the clinical features of Dubowitz syndrome in older patients. This deficiency was corrected in part by Hansen et al. (1995) who described long-term follow-up of one of the first patients to be diagnosed with Dubowitz syndrome (Grosse et al., 1971). ... Ahmad et al. (1999) reported a girl with Dubowitz syndrome and a persistently low cholesterol level. ... A 7-dehydrocholesterol level was normal, ruling out Smith-Lemli-Opitz syndrome (270400). Plasma sterol analysis by GC/MS was also normal.
Dubowitz syndrome (DS) is a rare multiple congenital syndrome characterized primarly by growth retardation, microcephaly, distinctive facial dysmorphism, cutaneous eczema, a mild to severe intellectual deficit and genital abnormalities. ... Etiology The etiology of Dubowitz syndrome has not been evidently elucidated. ... Differential diagnosis Differential diagnosis includes fetal alcohol syndrome, Bloom syndrome, LIG4 syndrome and Fanconi anemia (see these terms). ... Genetic counseling In some families, Dubowitz syndrome appears to have an autosomal recessive transmission. However, it has been suggested recently that Dubowitz syndrome is a microdeletion/microduplication syndrome rather than an autosomal disorder.
It is characterized by advanced osseous maturation and distinctive craniofacial, skeletal and neurological abnormalities. [1] It is similar to Sotos syndrome and is classified as an overgrowth syndrome . ... "NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes" . ... In Congenital Malformation Syndromes. New York: Chapman and Hall Medical, 1995, pp. 267-280. ^ NSD1 Mutations Are the Major Cause of Sotos Syndrome and Occur in Some Cases of Weaver Syndrome but Are Rare in Other Overgrowth Phenotypes Jenny Douglas, Sandra Hanks, I. ... "Weaver syndrome" . Gale Encyclopedia of Public Health . ... External links [ edit ] Classification D ICD - 10 : Q87.3 OMIM : 277590 MeSH : C536687 C562443, C536687 SNOMED CT : 63119004 External resources Orphanet : 3447 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome
Although there is phenotypic overlap between Weaver syndrome and Sotos syndrome (117550), distinguishing features of Weaver syndrome include broad forehead and face, ocular hypertelorism, prominent wide philtrum, micrognathia, deep horizontal chin groove, and deep-set nails. ... Sotos syndrome (117550), which shows considerable phenotypic overlap with Weaver syndrome, is caused by mutation in the NSD1 gene (601573) on chromosome 5q35. ... Possible phenotypic differences between the 2 syndromes pointed out by Opitz et al. (1998) were the following: the Sotos syndrome may be a cancer syndrome, whereas the Weaver syndrome is not (although a neuroblastoma had been reported in the latter disorder). In Sotos syndrome there is remarkably advanced dental maturation; this is rarely commented on in Weaver syndrome. In Weaver syndrome, there are more conspicuous contractures and a facial appearance that experts find convincingly different from that in Sotos syndrome.
Weaver syndrome (WVS) is a rare, multisystem disorder characterized by tall stature, a typical facial appearance (hypertelorism, retrognathia) and variable intellectual disability. ... Epidemiology Around 50 cases of Weaver syndrome have been reported to date. Precise prevalence and incidence rates are not available. ... Differential diagnosis The main differential diagnosis is Sotos syndrome (see this term) which has considerable overlap with WVS. Other disorders to consider include Beckwith-Wiedemann, Simpson-Golabi-Behmel, Malan overgrowth, tall stature-intellectual disability-facial dysmorphism and Marfan syndromes (see these terms). Antenatal diagnosis Prenatal diagnosis in at-risk pregnancies is possible.
Weaver syndrome is a condition that involves tall stature with or without a large head size (macrocephaly ), a variable degree of intellectual disability (usually mild), and characteristic facial features. ... Some affected individuals have abnormalities in the folds (gyri) of the brain, which can be seen by medical imaging; the relationship between these brain abnormalities and the intellectual disability associated with Weaver syndrome is unclear. Researchers suggest that people with Weaver syndrome may have an increased risk of developing cancer, in particular a slightly increased risk of developing a tumor called neuroblastoma in early childhood, but the small number of affected individuals makes it difficult to determine the exact risk. Frequency The prevalence of Weaver syndrome is unknown. About 50 affected individuals have been described in the medical literature. Causes Weaver syndrome is usually caused by mutations in the EZH2 gene. ... It is unclear how mutations in the EZH2 gene result in the abnormalities characteristic of Weaver syndrome. Learn more about the gene associated with Weaver syndrome EZH2 Inheritance Pattern This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Weaver syndrome is a rare condition that is characterized primarily by tall stature. ... Some studies also suggest that people affected by Weaver syndrome may have an increased risk of developing neuroblastoma . Weaver syndrome is usually caused by changes (mutations) in the EZH2 gene.
Laurence-Moon syndrome is a rare condition that affects many different parts of the body. Signs and symptoms vary but may include cerebellar ataxia ; eye abnormalities (primarily affecting the choroid and retina ); peripheral neuropathy; spastic paraplegia (progressive weakness and stiffness of the legs); intellectual disability; congenital (from birth) or childhood hypopituitarism ; and short stature. Laurence-Moon syndrome is caused by changes (mutations) in the PNPLA6 gene and is inherited in an autosomal recessive manner. Treatment is based on the signs and symptoms present in each person. Until recently, Laurence-Moon syndrome has been associated with Bardet-Biedl syndrome but newer research determined that they are separate conditions.
One such family has been reported. Description Laurence-Moon syndrome has a clinical presentation similar to that of Oliver-McFarlane syndrome (275400), including chorioretinopathy and pituitary dysfunction, but with childhood onset of ataxia, peripheral neuropathy, and spastic paraplegia and without trichomegaly. Historically, Laurence-Moon syndrome has been associated with Bardet-Biedl syndrome (see BBS, 209900) (summary by Hufnagel et al., 2015). ... Solis-Cohen and Weiss (1925) considered the disorder identical to Bardet and Biedl syndrome; they used the designation Laurence-Biedl syndrome. ... Hufnagel et al. (2015) considered the Laurence-Moon and Bardet-Biedl syndromes to be distinct, given the marked choroidal atrophy, hypopituitarism with short stature, early neurologic involvement, and the absence of polydactyly and renal disease in Laurence-Moon syndrome. ... Population Genetics Farag and Teebi (1988) concluded that both the Bardet-Biedl and the Laurence-Moon syndromes are increased in the Arab population of Kuwait.
A very rare genetic multisystemic disorder characterized by pituitary dysfunction, ataxia, peripheral neuropathy, spastic paraplegia, and chorioretinal dystrophy.
A number sign (#) is used with this entry because of evidence that Bamforth-Lazarus syndrome is caused by homozygous mutation in the FKHL15 gene (FOXE1; 602617) on chromosome 9q22. ... Buntincx et al. (1993) reported a female child with the same syndrome. Choanal atresia may have been responsible for the polyhydramnios which was observed in all 3 cases. ... Baris et al. (2006) reported a child, the daughter of consanguineous Turkish parents, with Bamforth-Lazarus syndrome who presented with congenital hypothyroidism, bilateral choanal atresia, cleft palate, and spiky hair but who was not athyreotic. ... In a girl, born of consanguineous Turkish parents, with Bamforth-Lazarus syndrome, Baris et al. (2006) identified homozygosity for a missense mutation affecting a highly conserved residue within the forkhead DNA-binding domain of FOXE1 (R102C; 602617.0003). Nomenclature Chatterjee (1998) suggested that the name of Lazarus, the senior author of the report by Bamforth et al. (1989), should be included with that of Bamforth, his resident, in the designation of the syndrome. HEENT - Spiky hair - Choanal atresia - Cleft palate - Bifid epiglottis Misc - Polyhydramnios pregnancy Endo - Athyroidal hypothyroidism Inheritance - Autosomal recessive ▲ Close
A very rare syndrome of congenital hypothyroidism characterized by thyroid dysgenesis (in most cases athyreosis), cleft palate and spiky hair, with or without choanal atresia, and bifid epiglottis. ... Epidemiology Only 8 patients from 6 families have been reported to date. Clinical description The syndrome is typically observed at birth with cleft palate, spiky hair and thyroid dysgenesis (in most cases athyreosis) leading to congenital hypothyroidism that manifests with lethargy, poor feeding, macroglossia, cold or mottled skin, persistent jaundice, and umbilical hernia. ... Porencephaly was also recently described in one case. Etiology Bamforth-Lazarus syndrome is due to homozygous loss-of-function missense mutations located within the forkhead domain of the FOXE1 gene (9q22), encoding thyroid transcription factor 2 (TTF-2). ... Differential diagnosis Differential diagnoses include other forms of syndromic hypothyroidism such as Johanson-Blizzard syndrome. ... Management and treatment Thyroid hormone replacement therapy is the standard treatment for those with Bamforth-Lazarus syndrome and should be started as soon as possible.
A number sign (#) is used with this entry because of evidence that ulnar-mammary syndrome (UMS) is caused by heterozygous mutation in the TBX3 gene (601621) on chromosome 12q24. ... They suggested that this finding supported the hypothesis of Lenz (1980) that the split hand with aplasia of the ulna syndrome is the same entity as the ulnar-mammary syndrome. ... In a Japanese mother and her 2 sons with ulnar-mammary syndrome, Sasaki et al. (2002) identified heterozygosity for a nonsense mutation in the TBX3 gene (K273X; 601621.0003). ... In a boy and his mother with ulnar-mammary syndrome, Linden et al. (2009) identified heterozygosity for a nonsense mutation in the TBX3 gene (Q331X; 601621.0005). ... In twin brothers and their father with ulnar-mammary syndrome, Tanteles et al. (2017) identified heterozygosity for a nonsense mutation in the TBX3 gene (Q475X; 601621.0006).
Ulnar-mammary syndrome (UMS) is a rare developmental disorder characterized by ulnar defects, mammary and apocrine gland hypoplasia and genital anomalies.
Saraux et al. (1963) described 2 sisters with Goldenhar syndrome, born of healthy, unrelated parents. ... Choong et al. (2003) reported a male infant, born of nonconsanguineous parents, who had clinical features of Goldenhar syndrome and the cri-du-chat syndrome (123450). ... The association of Goldenhar syndrome and cri-du-chat syndrome in this patient suggested that the chromosome 5p14 locus may harbor a gene implicated with Goldenhar syndrome. ... Descartes (2006) noted that Ladekarl (1968) had reported a patient with features of Goldenhar syndrome and cri-du-chat syndrome associated with a 5q deletion. ... The authors suggested that, at least for these 3 families, autosomal dominant OAVS is genetically distinct from the clinically similar syndromes caused by mutation in those 2 genes, branchiootorenal syndrome (113650) and Townes-Brocks syndrome (107480), respectively.
A rare congenital malformation syndrome, most commonly presenting with hemifacial microsomia associated with ear and/or eye malformations and vertebral anomalies of variable severity. ... Differential diagnosis Differential diagnosis includes syndromes associated with microtia and mandibular hypoplasia, including Treacher Collins syndrome, Townes-Brocks syndrome, CHARGE syndrome, branchio-oto-renal spectrum disorders, mandibulofacial dysostosis with microcephaly, and Wildervanck syndrome.
These conditions include hemifacial microsomia (when only one side of the face is affected), Goldenhar syndrome (hemifacial microsomia and noncancerous (benign) growths in the eye called epibulbar dermoids), and others, such as first and second branchial arch syndrome, otomandibular dysostosis, facio-auriculo-vertebral syndrome and lateral facial dysplasia.
First arch syndrome Specialty Medical genetics First arch syndromes are congenital defects caused by a failure of neural crest cells to migrate into the first pharyngeal arch . [1] They can produce facial anomalies . Examples of first arch syndromes include Treacher Collins syndrome and Pierre Robin syndrome .
Pfeiffer syndrome is a disorder that affects the development of the bones in the skull, hands and feet. ... It is caused by mutations in the FGFR1 or FGFR2 genes and is inherited in an autosomal dominant manner. Pfeiffer syndrome is divided into 3 subtypes ( type 1 , type 2 and type 3 ) based on the presence and severity of specific features.
Most of the affected patients show various other associated manifestations. Epidemiology Pfeiffer syndrome (PS) birth prevalence is 1/100,000. ... Patients meeting criteria for a clinical diagnosis of Pfeiffer syndrome without a mutation in FGFR1 or FGFR2 are estimated to be as high as 21%. ... Differential diagnosis Differential diagnoses include other acrocephalosyndactyly syndromes (Apert, Crouzon, Carpenter, Saethre-Chotzen, Waardenburg) and other syndromic forms of craniosynostosis such as Jackson-Weiss, Muenke and Antley-Bixler syndromes and Cutis gyrata-acanthosis nigricans-craniosynostosis. ... Inheritance from an affected parent in an autosomal dominant manner is reported in less severe cases of Pfeiffer syndrome. PS shows complete penetrance, although the expressivity is variable.
Pfeiffer syndrome is a genetic disorder characterized by the premature fusion of certain skull bones (craniosynostosis). ... More than half of all children with Pfeiffer syndrome have hearing loss; dental problems are also common. ... Type 1, also known as classic Pfeiffer syndrome, has symptoms as described above. Most individuals with type 1 Pfeiffer syndrome have normal intelligence and a normal life span. ... Causes Pfeiffer syndrome is most commonly caused by mutations in the FGFR2 gene.
Kroczek et al. (1986) described Kleblattschaedel in association with Pfeiffer syndrome. Rasmussen and Frias (1988) described a girl with severe manifestations of Pfeiffer syndrome. ... In a study of sporadic cases of Crouzon syndrome and Pfeiffer syndrome, Glaser et al. (2000) used 4 intragenic polymorphisms to screen a total of 41 families. ... Advanced paternal age was noted for the fathers of patients with Crouzon syndrome or Pfeiffer syndrome, compared with the fathers of control individuals (34.50 +/- 7.65 years vs 30.45 +/- 1.28 years, P less than 0.01). ... The patient had cloverleaf skull deformity as well as the other typical ocular, hand, and foot anomalies seen in Pfeiffer syndrome. Missense mutations at codon 290 of FGFR2 had been reported previously in Crouzon syndrome, but not in Pfeiffer syndrome. ... The same mutation had been reported by Schaefer et al. (1998) in a case of Pfeiffer syndrome with overlapping features of Antley-Bixler syndrome (207410).
Pfeiffer syndrome type 2 (PS2) is a frequent and severe type of Pfeiffer syndrome (PS; see this term), characterized by cloverleaf skull, severe associated functional disorders, and hand/foot and elbow/knee abnormalities. ... FGFR2 mutations are most commonly detected (69% in one study) followed by FGFR1 (8%). Genetic counseling Pfeiffer syndrome follows an autosomal dominant pattern of inheritance but is generally caused by de novo mutations, especially in the severe forms of the syndrome.
Pfeiffer syndrome type 1 (PS1) is a mild to moderately severe type of Pfeiffer syndrome (PS; see this term), characterized by bicoronal craniosynostosis, variable finger and toe malformations, and in most cases, normal intellectual development. ... Etiology PS1 is most commonly caused by mutations in the FGFR2 gene (10q25.3-q26), and in rare instances by mutations in the FGFR1 gene (8p11.23-p11.22). Genetic counseling Pfeiffer syndrome follows an autosomal dominant pattern of inheritance, observed primarily in non-severe cases compatible with long-term survival. ... Prognosis Due to decreased need for extensive corrective surgery and essentially normal intellectual development, overall prognosis in PS1 is much more favorable than in Pfeiffer syndrome type 2 or type 3.
Pfeiffer syndrome type 3 (PS3) is a severe type of Pfeiffer syndrome (PS; see this term), characterized by bicoronal craniosynostosis, severe associated functional disorders, and hand, foot and elbow abnormalities. ... Genetic counseling PS follows an autosomal dominant pattern of inheritance but is generally caused by de novo mutations, especially in the severe forms of the syndrome. Genetic counseling should be provided to affected families.
A rare non-syndromic syndactyly characterized by a distinctive combination of syndactyly and polydactyly, generally affecting the 3rd and 4th fingers and the 4th and 5th toes, bilaterally, with partial or complete reduplication of a digital ray within the syndactylous web.
For a general phenotypic description and a discussion of genetic heterogeneity of synpolydactyly, see SPD1 (186000). Clinical Features Malik et al. (2006) reported a large Pakistani kindred in which 16 members over 6 generations had synpolydactyly. The cardinal features included osseous fusion of the third and fourth fingers and postaxial synpolydactyly of toes. Variable features included cutaneous webbing, symphalangism, abnormal metacarpals, clinodactyly, and camptodactyly. Inheritance The transmission pattern of synpolydactyly in the Pakistani family reported by Malik et al. (2006) was consistent with autosomal dominant inheritance.
Zakany and Duboule (1996) suggested that this syndrome, which is associated with a subtle mutation in human HOXD13, may involve the loss of function of several HOXD genes.
Clinical Features De Smet et al. (1996) reported a Belgian family in which a father and daughter had a syndrome with metatarsal fusion, metacarpal fusion, and synpolydactyly between the third and fourth fingers. ... The authors suggested that earlier reported cases of Cenani-Lenz syndrome in sibs of normal parents may have been due to gonadal mosaicism or that Cenani syndactyly is genetically heterogeneous with autosomal recessive and autosomal dominant forms. ... Debeer et al. (1998, 1998) referred to the syndrome in their family as a complex type of '3/3-prime/4 synpolydactyly associated with metacarpal and metatarsal synostoses.' ... The authors noted that the entire FBLN1 gene is deleted in the chromosome 22q13.3 deletion syndrome (606232). INHERITANCE - Autosomal dominant SKELETAL Hands - Syndactyly (often 3rd and 4th digits) - Synpolydactyly (3rd or 4th digits) - Metacarpal synostosis (4th and 5th digits) - Symmetric, bilateral malformations Feet - Syndactyly (2nd, 3rd, 4th digits) - Metatarsal synostosis (3rd and 4th digits) - Symmetric, bilateral malformations MISCELLANEOUS - Associated with a balanced translocation t(12,22)(p11.2,q13.3) MOLECULAR BASIS - Caused by translocation disrupting the fibulin-1 gene (FBLN1, 135820 ) ▲ Close
Summary Clinical characteristics. Baller-Gerold syndrome (BGS) can be suspected at birth in an infant with craniosynostosis and upper limb abnormality. ... Genetic counseling. Baller-Gerold syndrome is inherited in an autosomal recessive manner. ... Therefore, individuals with Baller-Gerold syndrome with symptoms suggestive of cancer should have prompt evaluation. ... Prevalence The prevalence of Baller-Gerold syndrome is unknown; it is probably less than 1:1,000,000 [Mo et al 2018]. Differential Diagnosis The major differential diagnosis for Baller-Gerold syndrome (BGS) comprises the allelic disorders Rothmund-Thomson syndrome and RAPADILINO syndrome (OMIM 266280).
Cases reported as Baller-Gerold syndrome have phenotypic overlap with several other disorders, including Saethre-Chotzen syndrome (SCS; 101400). ... Huson et al. (1990) suggested that cases diagnosed as having Baller-Gerold syndrome should have cytogenetic analysis and, conversely, that known Roberts syndrome survivors should be reviewed for signs of craniostenosis. ... Cohen and Toriello (1996) likewise raised the question 'Is there a Baller-Gerold syndrome?' As outlined earlier, phenotypic overlap of Baller-Gerold syndrome with several other syndromes required narrowing the definition of Baller-Gerold syndrome. ... The father, who also carried the mutation, had very mild features of Saethre-Chotzen syndrome. Since mutations in the TWIST gene are typically associated with SCS, the authors suggested that some cases of Baller-Gerold syndrome should be reclassified as a heterogeneous form of Saethre-Chotzen syndrome. de Oliveira et al. (2005) reported 3 patients with a history of fetal exposure to sodium valproate who were born with metopic synostosis and upper limb malformations similar to Baller-Gerold syndrome. ... Molecular Genetics Van Maldergem et al. (2006) pointed out the clinical overlap between the BGS and Rothmund-Thomson syndrome (RTS; 268400) and RAPADILINO syndrome (266280).
Baller-Gerold syndrome is characterized by the association of coronal craniosynostosis with radial ray anomalies (oligodactyly, aplasia or hypoplasia of the thumb, aplasia or hypoplasia of the radius). ... Patients have a predisposition to cancer, in particular osteosarcoma. Etiology Baller-Gerold syndrome is secondary to mutations of the RECQL4 gene (8q24.3). ... Differential diagnosis The principal differential diagnoses include Rothmund-Thomson syndrome (RTS) and RAPADILINO syndrome, also secondary to mutations of the RECQL4 gene (see these terms). ... Other differential diagnoses include Roberts syndrome and Fanconi anemia, which are frequently associated with radial ray anomalies but rarely with craniosynostosis, and Saethre-Chotzen syndrome which is characterized by coronal craniosynostosis usually without radial ray anomalies (see these terms). ... Genetic counseling Baller-Gerold syndrome is inherited in an autosomal recessive manner.
Baller-Gerold syndrome (BGS) is a rare condition primarily affecting the way the bones of the skull and limbs grow. ... Treatment may include surgery for craniosynostosis or for reconstruction of the arm and hand. The symptoms of Baller-Gerold syndrome overlap with features of Rothmund-Thomson syndrome and RAPADILINO syndrome which are also caused by the RECQL4 gene.
Baller-Gerold syndrome is a rare condition characterized by the premature fusion of certain skull bones (craniosynostosis) and abnormalities of bones in the arms and hands. ... The varied signs and symptoms of Baller-Gerold syndrome overlap with features of other disorders, namely Rothmund-Thomson syndrome and RAPADILINO syndrome. These syndromes are also characterized by radial ray defects, skeletal abnormalities, and slow growth. ... Based on these similarities, researchers are investigating whether Baller-Gerold syndrome, Rothmund-Thomson syndrome, and RAPADILINO syndrome are separate disorders or part of a single syndrome with overlapping signs and symptoms. Frequency The prevalence of Baller-Gerold syndrome is unknown, but this rare condition probably affects fewer than 1 per million people.
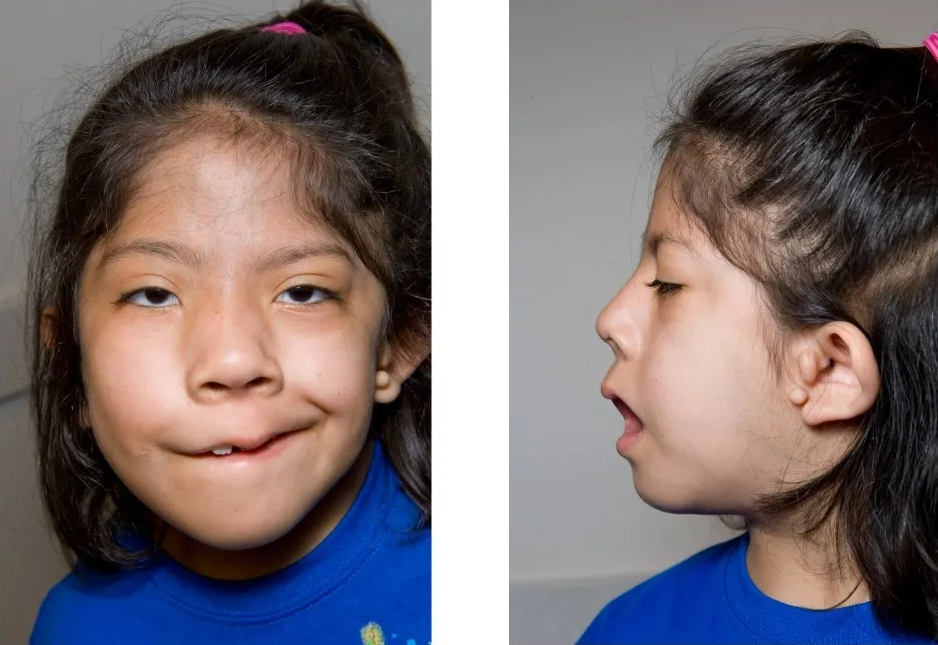
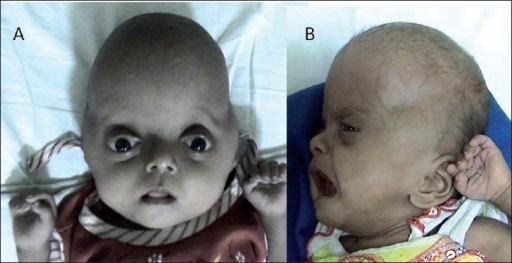
Langer–Giedion syndrome Other names Deletion 8q24.1, monosomy 8q24.1, trichorhinophalangeal syndrome type II (TRPS2), Langer-Giedion chromosome region (LGCR) [1] [2] A person showing the typical features of Langer-Giedion syndrome Specialty Medical genetics Langer–Giedion syndrome ( LGS ) is a very uncommon autosomal dominant genetic disorder caused by a deletion of a small section of material on chromosome 8 . ... Contents 1 Signs and symptoms 2 Cause 3 Diagnosis 4 Treatment 5 See also 6 References 7 External links Signs and symptoms [ edit ] The features associated with this condition include: mild to moderate learning difficulties, short stature, unique facial features, small head and skeletal abnormalities including bony growths projecting from the surfaces of bones. [3] Typically, individuals with Langer–Giedion syndrome have fine scalp hair, ears that may be large or prominent, broad eyebrows, deep-set eyes, a bulbous nose, a long narrow upper lip and missing teeth. [ citation needed ] The right foot of a person with Langer–Giedion syndrome showing the characteristic features Hands of a person with Langer–Giedion syndrome showing the characteristic short fingers Cause [ edit ] The syndrome occurs when a small piece of chromosome 8's long arm, which contains a number of genes , is missing. ... Nowadays, it is a common practice to run an aCGH (array chromosome hybridization genome) study on peripheral blood of the patient, in order to delineate the extent of the loss of the genomic area, and the deleted genes. [4] Treatment [ edit ] While no genetic syndrome is capable of being cured, treatments are available for some symptoms. ... PMID 18478595 . ^ Devidayal; Marwaha RK (February 2006). "Langer-Giedion Syndrome" (PDF) . Indian Pediatrics . 43 (2): 174–175. PMID 16528117 . ^ http://www.omim.org/entry/150230 External links [ edit ] Classification D ICD - 10 : ICD-10: Q87.8 OMIM : 150230 MeSH : D015826 DiseasesDB : 31949 External resources Orphanet : 502 Trichorhinophalangeal syndrome type 2 at NIH 's Office of Rare Diseases v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions )
A number sign (#) is used with this entry because trichorhinophalangeal syndrome type II (TRPS2), also known as Langer-Giedion syndrome, is a contiguous gene syndrome on 8q24.1, involving loss of functional copies of the TRPS1 (604386) and EXT1 (608177) genes. ... See also Cornelia de Lange syndrome-4 (CDLS4; 614701), caused by mutation in the RAD21 gene (606462), which lies on 8q24 between TRPS1 and EXT1. ... Morioka et al. (1999) described a patient with Langer-Giedion syndrome associated with submucous cleft palate. ... They raised the question of whether type I trichorhinophalangeal syndrome may be caused by mutation at the same locus or region. ... They concluded that the Langer-Giedion syndrome is not due to pleiotropic effects of mutations in a single gene, but that it is a true contiguous gene syndrome.
Trichorhinophalangeal syndrome type II (TRPS II) is a condition that causes bone and joint malformations; distinctive facial features; intellectual disability; and abnormalities of the skin, hair, teeth, sweat glands, and nails. ... TRPS II is often described as a contiguous gene deletion syndrome because it results from the loss of several neighboring genes. ... This condition, called trichorhinophalangeal syndrome type I (TRPS I), features similar bone, joint, skin, and facial characteristics as TRPS II. ... Learn more about the genes and chromosome associated with Trichorhinophalangeal syndrome type II EXT1 RAD21 TRPS1 chromosome 8 Inheritance Pattern Most cases of TRPS II are not inherited, but occur as random events during the formation of reproductive cells (eggs or sperm) in a parent of an affected individual.
A very rare, genetic, multiple congenital anomaly disorder characterized by bone abnormalities, distinctive facial features, multiple exostoses, and intellectual disability.