-
Adermatoglyphia
Wikipedia
It results in a shortened form of the skin-specific protein . [5] The heterozygous expression of the mutation suggests an autosomal dominant mode of inheritance. [6] The Swiss patient, and eight of her relatives who also had the mutation, all had "flat finger pads and a reduced number of sweat glands in the hands". [7] Other conditions can cause a lack of fingerprints, but unlike them, adermatoglyphia has no side effects. [8] Mutations in helicases are involved in other rare genetic diseases, such as Werner syndrome . [9] References [ edit ] ^ Reference, Genetics Home. ... External links [ edit ] The dictionary definition of adermatoglyphia at Wiktionary Classification D ICD - 10 : Q82.8 OMIM : 136000 MeSH : C565010 External resources Orphanet : 289465 v t e Metabolic disease : DNA replication and DNA repair-deficiency disorder DNA replication Separation/initiation: RNASEH2A Aicardi–Goutières syndrome 4 Termination/ telomerase : DKC1 Dyskeratosis congenita DNA repair Nucleotide excision repair Cockayne syndrome / DeSanctis–Cacchione syndrome Thymine dimer Xeroderma pigmentosum IBIDS syndrome MSI / DNA mismatch repair Hereditary nonpolyposis colorectal cancer Muir–Torre syndrome Mismatch repair cancer syndrome MRN complex Ataxia telangiectasia Nijmegen breakage syndrome Other RecQ helicase Bloom syndrome Werner syndrome Rothmund–Thomson syndrome / Rapadilino syndrome Fanconi anemia Li-Fraumeni syndrome Severe combined immunodeficiency This cutaneous condition article is a stub .

-
Cd25 Deficiency
Wikipedia
The mutations cause expression of a defective α chain or complete absence thereof, an essential part of high-affinity interleukin-2 (IL-2) receptors. The result is a syndrome described as IPEX -like [1] or a SCID . [2] In one patient, deficiency of CD25 on CD4+ lymphocytes caused significantly impaired sensitivity to IL-2. ... Greatly reduced IL-10 secretion compared to healthy humans results in a syndrome comparable to IPEX syndrome, a type of autoimmunity which is caused by FoxP3 transcription factor dysfunction. ... "CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked–like syndrome, and defective IL-10 expression from CD4 lymphocytes". ... External links [ edit ] Classification D ICD - 10 : D81.2 OMIM : 606367 MeSH : C565232 External resources Orphanet : 169100 v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency

-
Onychauxis
Wikipedia
External links [ edit ] Classification D ICD - 10 : L60.2 , Q84.5 ICD - 9-CM : 703.8 , 757.5 DiseasesDB : 32084 v t e Disorders of skin appendages Nail thickness: Onychogryphosis Onychauxis color: Beau's lines Yellow nail syndrome Leukonychia Azure lunula shape: Koilonychia Nail clubbing behavior: Onychotillomania Onychophagia other: Ingrown nail Anonychia ungrouped: Paronychia Acute Chronic Chevron nail Congenital onychodysplasia of the index fingers Green nails Half and half nails Hangnail Hapalonychia Hook nail Ingrown nail Lichen planus of the nails Longitudinal erythronychia Malalignment of the nail plate Median nail dystrophy Mees' lines Melanonychia Muehrcke's lines Nail–patella syndrome Onychoatrophy Onycholysis Onychomadesis Onychomatricoma Onychomycosis Onychophosis Onychoptosis defluvium Onychorrhexis Onychoschizia Platonychia Pincer nails Plummer's nail Psoriatic nails Pterygium inversum unguis Pterygium unguis Purpura of the nail bed Racquet nail Red lunulae Shell nail syndrome Splinter hemorrhage Spotted lunulae Staining of the nail plate Stippled nails Subungual hematoma Terry's nails Twenty-nail dystrophy Hair Hair loss / Baldness noncicatricial alopecia : Alopecia areata totalis universalis Ophiasis Androgenic alopecia (male-pattern baldness) Hypotrichosis Telogen effluvium Traction alopecia Lichen planopilaris Trichorrhexis nodosa Alopecia neoplastica Anagen effluvium Alopecia mucinosa cicatricial alopecia : Pseudopelade of Brocq Central centrifugal cicatricial alopecia Pressure alopecia Traumatic alopecia Tumor alopecia Hot comb alopecia Perifolliculitis capitis abscedens et suffodiens Graham-Little syndrome Folliculitis decalvans ungrouped: Triangular alopecia Frontal fibrosing alopecia Marie Unna hereditary hypotrichosis Hypertrichosis Hirsutism Acquired localised generalised patterned Congenital generalised localised X-linked Prepubertal Acneiform eruption Acne Acne vulgaris Acne conglobata Acne miliaris necrotica Tropical acne Infantile acne / Neonatal acne Excoriated acne Acne fulminans Acne medicamentosa (e.g., steroid acne ) Halogen acne Iododerma Bromoderma Chloracne Oil acne Tar acne Acne cosmetica Occupational acne Acne aestivalis Acne keloidalis nuchae Acne mechanica Acne with facial edema Pomade acne Acne necrotica Blackhead Lupus miliaris disseminatus faciei Rosacea Perioral dermatitis Granulomatous perioral dermatitis Phymatous rosacea Rhinophyma Blepharophyma Gnathophyma Metophyma Otophyma Papulopustular rosacea Lupoid rosacea Erythrotelangiectatic rosacea Glandular rosacea Gram-negative rosacea Steroid rosacea Ocular rosacea Persistent edema of rosacea Rosacea conglobata variants Periorificial dermatitis Pyoderma faciale Ungrouped Granulomatous facial dermatitis Idiopathic facial aseptic granuloma Periorbital dermatitis SAPHO syndrome Follicular cysts " Sebaceous cyst " Epidermoid cyst Trichilemmal cyst Steatocystoma simplex multiplex Milia Inflammation Folliculitis Folliculitis nares perforans Tufted folliculitis Pseudofolliculitis barbae Hidradenitis Hidradenitis suppurativa Recurrent palmoplantar hidradenitis Neutrophilic eccrine hidradenitis Ungrouped Acrokeratosis paraneoplastica of Bazex Acroosteolysis Bubble hair deformity Disseminate and recurrent infundibulofolliculitis Erosive pustular dermatitis of the scalp Erythromelanosis follicularis faciei et colli Hair casts Hair follicle nevus Intermittent hair–follicle dystrophy Keratosis pilaris atropicans Kinking hair Koenen's tumor Lichen planopilaris Lichen spinulosus Loose anagen syndrome Menkes kinky hair syndrome Monilethrix Parakeratosis pustulosa Pili ( Pili annulati Pili bifurcati Pili multigemini Pili pseudoannulati Pili torti ) Pityriasis amiantacea Plica neuropathica Poliosis Rubinstein–Taybi syndrome Setleis syndrome Traumatic anserine folliculosis Trichomegaly Trichomycosis axillaris Trichorrhexis ( Trichorrhexis invaginata Trichorrhexis nodosa ) Trichostasis spinulosa Uncombable hair syndrome Wooly hair nevus Sweat glands Eccrine Miliaria Colloid milium Miliaria crystalline Miliaria profunda Miliaria pustulosa Miliaria rubra Occlusion miliaria Postmiliarial hypohidrosis Granulosis rubra nasi Ross’ syndrome Anhidrosis Hyperhidrosis Generalized Gustatory Palmoplantar Apocrine Body odor Chromhidrosis Fox–Fordyce disease Sebaceous Sebaceous hyperplasia v t e Congenital malformations and deformations of skin appendages Nail disease Anonychia Leukonychia Pachyonychia congenita / Onychauxis Koilonychia Hair disease hypotrichosis /abnormalities: keratin disease Monilethrix IBIDS syndrome Sabinas brittle hair syndrome Pili annulati Pili torti Uncombable hair syndrome Björnstad syndrome Giant axonal neuropathy with curly hair hypertrichosis : Zimmermann–Laband syndrome This condition of the skin appendages article is a stub .

-
Hair Disease
Wikipedia
See also [ edit ] List of cutaneous conditions References [ edit ] External links [ edit ] Classification D ICD - 10 : L63 - L73 , Q84.1 - Q84.2 ICD - 9-CM : 704 , 757.4 MeSH : D006201 https://www.nlm.nih.gov/medlineplus/hairdiseasesandhairloss.html v t e Disorders of skin appendages Nail thickness: Onychogryphosis Onychauxis color: Beau's lines Yellow nail syndrome Leukonychia Azure lunula shape: Koilonychia Nail clubbing behavior: Onychotillomania Onychophagia other: Ingrown nail Anonychia ungrouped: Paronychia Acute Chronic Chevron nail Congenital onychodysplasia of the index fingers Green nails Half and half nails Hangnail Hapalonychia Hook nail Ingrown nail Lichen planus of the nails Longitudinal erythronychia Malalignment of the nail plate Median nail dystrophy Mees' lines Melanonychia Muehrcke's lines Nail–patella syndrome Onychoatrophy Onycholysis Onychomadesis Onychomatricoma Onychomycosis Onychophosis Onychoptosis defluvium Onychorrhexis Onychoschizia Platonychia Pincer nails Plummer's nail Psoriatic nails Pterygium inversum unguis Pterygium unguis Purpura of the nail bed Racquet nail Red lunulae Shell nail syndrome Splinter hemorrhage Spotted lunulae Staining of the nail plate Stippled nails Subungual hematoma Terry's nails Twenty-nail dystrophy Hair Hair loss / Baldness noncicatricial alopecia : Alopecia areata totalis universalis Ophiasis Androgenic alopecia (male-pattern baldness) Hypotrichosis Telogen effluvium Traction alopecia Lichen planopilaris Trichorrhexis nodosa Alopecia neoplastica Anagen effluvium Alopecia mucinosa cicatricial alopecia : Pseudopelade of Brocq Central centrifugal cicatricial alopecia Pressure alopecia Traumatic alopecia Tumor alopecia Hot comb alopecia Perifolliculitis capitis abscedens et suffodiens Graham-Little syndrome Folliculitis decalvans ungrouped: Triangular alopecia Frontal fibrosing alopecia Marie Unna hereditary hypotrichosis Hypertrichosis Hirsutism Acquired localised generalised patterned Congenital generalised localised X-linked Prepubertal Acneiform eruption Acne Acne vulgaris Acne conglobata Acne miliaris necrotica Tropical acne Infantile acne / Neonatal acne Excoriated acne Acne fulminans Acne medicamentosa (e.g., steroid acne ) Halogen acne Iododerma Bromoderma Chloracne Oil acne Tar acne Acne cosmetica Occupational acne Acne aestivalis Acne keloidalis nuchae Acne mechanica Acne with facial edema Pomade acne Acne necrotica Blackhead Lupus miliaris disseminatus faciei Rosacea Perioral dermatitis Granulomatous perioral dermatitis Phymatous rosacea Rhinophyma Blepharophyma Gnathophyma Metophyma Otophyma Papulopustular rosacea Lupoid rosacea Erythrotelangiectatic rosacea Glandular rosacea Gram-negative rosacea Steroid rosacea Ocular rosacea Persistent edema of rosacea Rosacea conglobata variants Periorificial dermatitis Pyoderma faciale Ungrouped Granulomatous facial dermatitis Idiopathic facial aseptic granuloma Periorbital dermatitis SAPHO syndrome Follicular cysts " Sebaceous cyst " Epidermoid cyst Trichilemmal cyst Steatocystoma simplex multiplex Milia Inflammation Folliculitis Folliculitis nares perforans Tufted folliculitis Pseudofolliculitis barbae Hidradenitis Hidradenitis suppurativa Recurrent palmoplantar hidradenitis Neutrophilic eccrine hidradenitis Ungrouped Acrokeratosis paraneoplastica of Bazex Acroosteolysis Bubble hair deformity Disseminate and recurrent infundibulofolliculitis Erosive pustular dermatitis of the scalp Erythromelanosis follicularis faciei et colli Hair casts Hair follicle nevus Intermittent hair–follicle dystrophy Keratosis pilaris atropicans Kinking hair Koenen's tumor Lichen planopilaris Lichen spinulosus Loose anagen syndrome Menkes kinky hair syndrome Monilethrix Parakeratosis pustulosa Pili ( Pili annulati Pili bifurcati Pili multigemini Pili pseudoannulati Pili torti ) Pityriasis amiantacea Plica neuropathica Poliosis Rubinstein–Taybi syndrome Setleis syndrome Traumatic anserine folliculosis Trichomegaly Trichomycosis axillaris Trichorrhexis ( Trichorrhexis invaginata Trichorrhexis nodosa ) Trichostasis spinulosa Uncombable hair syndrome Wooly hair nevus Sweat glands Eccrine Miliaria Colloid milium Miliaria crystalline Miliaria profunda Miliaria pustulosa Miliaria rubra Occlusion miliaria Postmiliarial hypohidrosis Granulosis rubra nasi Ross’ syndrome Anhidrosis Hyperhidrosis Generalized Gustatory Palmoplantar Apocrine Body odor Chromhidrosis Fox–Fordyce disease Sebaceous Sebaceous hyperplasia v t e Congenital malformations and deformations of skin appendages Nail disease Anonychia Leukonychia Pachyonychia congenita / Onychauxis Koilonychia Hair disease hypotrichosis /abnormalities: keratin disease Monilethrix IBIDS syndrome Sabinas brittle hair syndrome Pili annulati Pili torti Uncombable hair syndrome Björnstad syndrome Giant axonal neuropathy with curly hair hypertrichosis : Zimmermann–Laband syndrome This condition of the skin appendages article is a stub .ARG1, SHOC2, EIF2AK4, KRT71, KRT75, GABPA, NR3C1, NFE2L2, THY1, DSP, CP, LPAR6, OVOL2, KRT74, KRT80, DSG4, HEPHL1

-
Familial Cold Autoinflammatory Syndrome 1
Omim
A number sign (#) is used with this entry because familial cold autoinflammatory syndrome-1 (FCAS1) is caused by heterozygous mutation in the NLRP3 gene (606416), encoding cryopyrin, on chromosome 1q44. Description Familial cold autoinflammatory syndrome is characterized clinically by recurrent attacks of a maculopapular rash associated with arthralgias, myalgias, fever and chills, and swelling of the extremities after exposure to cold. ... Rarely, some patients may also develop late-onset renal amyloidosis (Hoffman et al., 2000). Overlapping syndromes also caused by mutation in the NLRP3 gene include Muckle-Wells syndrome (CAPS2; 191900), which has a high frequency of amyloidosis and late-onset sensorineural deafness, and chronic neurologic cutaneous and articular syndrome (CINCA, CAPS3; 607115), which shows earlier onset and a more severe phenotype. ... Jung et al. (1996) had also suggested that familial cold urticaria and Muckle-Wells syndrome are probably allelic. Molecular Genetics In 3 unrelated families with familial cold autoinflammatory syndrome, Hoffman et al. (2001) found 3 missense mutations in exon 3 of the CIAS1 gene (606416.0001-606416.0003). The families had been described by Shepard (1971), Vlagopoulos et al. (1975), and Wanderer (1979), respectively. In 1 family with Muckle-Wells syndrome, Hoffman et al. (2001) found a mutation in the CIAS1 gene (606416.0004), demonstrating that these 2 syndromes are indeed allelic.
-
Nezelof Syndrome
Wikipedia
Nezelof syndrome Other names Thymic dysplasia with normal immunoglobulins [1] : 85 Autosomal recessive is the manner in which this condition is inherited Specialty Immunology Symptoms Hepatosplenomegaly [2] Causes Currently unknown [3] Diagnostic method Blood test [3] [4] Treatment Antimicrobial therapy, IV immunoglobulin [5] Nezelof syndrome is an autosomal recessive [6] congenital immunodeficiency condition due to underdevelopment of the thymus . ... Ribonucleotide reductase catalyzes the formation of deoxyribonucleotides from ribonucleotides , thus, DNA replication is inhibited. [ medical citation needed ] Contents 1 Symptoms and signs 2 Cause 3 Mechanism 4 Diagnosis 4.1 Differential diagnosis 5 Treatment 6 See also 7 References 8 Further reading 9 External links Symptoms and signs [ edit ] This condition causes severe infections. it is characterized by elevated immunoglobulins that function poorly. [7] [8] Other symptoms are: [2] Bronchiectasis Hepatosplenomegaly Pyoderma Emphysema Diarrhea Cause [ edit ] Genetically speaking, Nezelof syndrome is autosomal recessive. the condition is thought to be a variation of severe combined immunodeficiency (SCID) [8] However, the precise cause of Nezelof syndrome remains uncertain [3] Mechanism [ edit ] In the mechanism of this condition, one first finds that the normal function of the thymus has it being important in T-cell development and release into the body's blood circulation [9] Hassal's corpuscles [10] absence in thymus(atrophy) has an effect on T-cells. [3] Diagnosis [ edit ] Human Thymus The diagnosis of Nezelof syndrome will indicate a deficiency of T-cells , [11] additionally in ascertaining the condition the following is done: [3] [4] Blood test ( B-cell will be normal) X-ray of thymus (atrophy present) Differential diagnosis [ edit ] The differential diagnosis for this condition consists of acquired immune deficiency syndrome and severe combined immunodeficiency syndrome [3] [8] Treatment [ edit ] Bone marrow for transplant In terms of treatment for individuals with Nezelof syndrome, which was first characterized in 1964, [12] includes the following(how effective bone marrow transplant is uncertain [4] ) : Antimicrobial therapy [5] IV immunoglobulin [5] Bone marrow transplantation [5] Thymus transplantation [5] Thymus factors [5] See also [ edit ] List of radiographic findings associated with cutaneous conditions References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). ... Lippincott Williams & Wilkins. p. 504 . ISBN 9780781730556 . Nezelof syndrome diagnosis. ^ Nezelof, C.; Jammet, M. ... External links [ edit ] PubMed Classification D ICD - 10 : D81.4 ICD - 9-CM : 279.13 OMIM : 242700 MeSH : C536288 DiseasesDB : 29571 External resources Orphanet : 83471 Scholia has a topic profile for Nezelof syndrome . v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
-
Linburg–comstock Variation
Wikipedia
Linburg–Comstock variation Other names Linburg–Comstock syndrome Linburg and Comstock syndrome is seen as a tendinous connection (green) between flexor pollicis longus (purple) and flexor digitorum profundus (yellow). ... Flexor tenosynovitis is a common finding in the patients with Linburg–Comstock syndrome. Another hypothesis is that anatomical variations, which in this case is an additional tendon slip, may act as space-occupying lesions and potentially contribute to carpal tunnel syndrome . [5] Structure [ edit ] Development [ edit ] Linburg–Comstock variation and syndrome may result from phylogenetic differences between human and non-human primates . ... "Flexor tendon anomalies in a patient with carpal tunnel syndrome". Journal of Hand Surgery . 26 (4): 373–376. doi : 10.1054/jhsb.2001.0613 . ... "Operative treatment of Linburg–Comstock syndrome" . The Journal of Bone and Joint Surgery. ... External links [ edit ] Classification D v t e Diseases relating to the peripheral nervous system Mononeuropathy Arm median nerve Carpal tunnel syndrome Ape hand deformity ulnar nerve Ulnar nerve entrapment Froment's sign Ulnar tunnel syndrome Ulnar claw radial nerve Radial neuropathy Wrist drop Cheiralgia paresthetica long thoracic nerve Winged scapula Backpack palsy Leg lateral cutaneous nerve of thigh Meralgia paraesthetica tibial nerve Tarsal tunnel syndrome plantar nerve Morton's neuroma superior gluteal nerve Trendelenburg's sign sciatic nerve Piriformis syndrome Cranial nerves See Template:Cranial nerve disease Polyneuropathy and Polyradiculoneuropathy HMSN Charcot–Marie–Tooth disease Dejerine–Sottas disease Refsum's disease Hereditary spastic paraplegia Hereditary neuropathy with liability to pressure palsy Familial amyloid neuropathy Autoimmune and demyelinating disease Guillain–Barré syndrome Chronic inflammatory demyelinating polyneuropathy Radiculopathy and plexopathy Brachial plexus injury Thoracic outlet syndrome Phantom limb Other Alcoholic polyneuropathy Other General Complex regional pain syndrome Mononeuritis multiplex Peripheral neuropathy Neuralgia Nerve compression syndrome
-
Chronic Recurrent Multifocal Osteomyelitis
Wikipedia
This particular classification encompasses both hereditary types ( familial Mediterranean fever , mevalonate kinase deficiency , TNF receptor associated periodic syndrome , cryopyrin-associated periodic syndrome , Blau syndrome , pyogenic sterile arthritis , pyoderma gangrenosum and acne syndrome, CRMO) and multifactorial disorders ( Crohn's and Behçet's diseases ). ... Other experts found that "mutations in LPIN2 cause a syndromic form of chronic recurrent multifocal osteomyelitis known as Majeed syndrome , while mutations in pstpip2 cause a murine form of the disorder. ... That means out of six million, there will probably be 5 girls and 1 boy with the condition. Majeed syndrome [ edit ] Majeed syndrome is an autoinflammatory disorder consisting of CRMO, congenital dyserythropoietic anemia , and neutrophilic dermatosis. To date, two unrelated families with Majeed syndrome have been reported. Mutations in LPIN2 have been found in both families. ... The two brothers also had Sweet syndrome . The association of Sweet syndrome with chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anemia in this family suggests that these rare conditions may be interrelated. [3] Notes [ edit ] ^ El-Shanti, HI; Ferguson, PJ (September 2007).
-
Posterior Cord Syndrome
Wikipedia
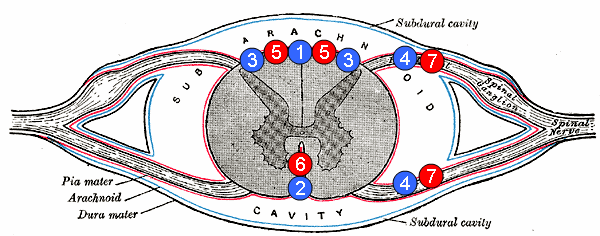
Human spinal cord disorder Posterior cord syndrome 5: posterior spinal arteries Posterior cord syndrome (PCS) , also known as posterior spinal artery syndrome (PSA), is a type of incomplete spinal cord injury . [1] PCS is the least commonly occurring of the six clinical spinal cord injury syndromes , with an incidence rate of less than 1%. ... "Incidence and Outcomes of Spinal Cord Injury Clinical Syndromes" . The Journal of Spinal Cord Medicine . ... "A case of Posterior Spinal Artery Syndrome in the Cervical Cord: A Review of the Clinicoradiological Literature" . ... "Cervical Posterior Spinal Artery Syndrome: A Case Report and Literature Review" . ... External links [ edit ] Classification D ICD - 10 : G95.1 ICD - 9-CM : 952.04 , 952.09 v t e Focal lesions of the spinal cord General Myelopathy Myelitis Spinal cord compression By location Brown-Séquard syndrome Posterior cord syndrome Anterior cord syndrome Central cord syndrome Cauda equina syndrome Other Polio Demyelinating disease Transverse myelitis Tropical spastic paraparesis Epidural abscess Syringomyelia Syringobulbia Morvan's syndrome Sensory ataxia Tabes dorsalis Abadie's sign Subacute combined degeneration of spinal cord Vascular myelopathy Anterior spinal artery syndrome Foix–Alajouanine syndrome
-
Tumor Lysis Syndrome
Wikipedia
Tumor lysis syndrome Other names TLS Specialty Oncology , hematology Tumor lysis syndrome is a group of metabolic abnormalities that can occur as a complication during the treatment of cancer , [1] where large amounts of tumor cells are killed off ( lysed ) at the same time by the treatment, releasing their contents into the bloodstream . ... Two common conditions related to excess uric acid , gout and uric acid nephrolithiasis , are not features of tumor lysis syndrome. Lactic acidosis . [5] [6] Pretreatment spontaneous tumor lysis syndrome . ... The important distinction between this syndrome and the post-chemotherapy syndrome is that spontaneous TLS is not associated with hyperphosphatemia. ... "Severe spontaneous acute tumor lysis syndrome and hypoglycemia in patient with germ cell tumor". ... S2CID 7889581 . ^ Cairo MS, Bishop M (October 2004). "Tumour lysis syndrome: new therapeutic strategies and classification" .
-
Haemophilia B
Wikipedia
This complex (in the coagulation pathway) will eventually activate factor X . [11] Diagnosis [ edit ] The diagnosis for haemophilia B can be done via the following tests/methods: [2] Coagulation screening test Bleeding scores Coagulation factor assays Differential diagnosis [ edit ] The differential diagnosis for this inherited condition is the following: haemophilia A , factor XI deficiency, von Willebrand disease , fibrinogen disorders and Bernard–Soulier syndrome [9] Treatment [ edit ] Treatment is given intermittently, when there is significant bleeding. ... External links [ edit ] Classification D ICD - 10 : D67 ICD - 9-CM : 286.1 OMIM : 306900 MeSH : D002836 DiseasesDB : 5561 External resources MedlinePlus : 000539 eMedicine : emerg/240 Patient UK : Haemophilia B Scholia has a topic profile for Haemophilia B . v t e Disorders of bleeding and clotting Coagulation · coagulopathy · Bleeding diathesis Clotting By cause Clotting factors Antithrombin III deficiency Protein C deficiency Activated protein C resistance Protein S deficiency Factor V Leiden Prothrombin G20210A Platelets Sticky platelet syndrome Thrombocytosis Essential thrombocythaemia DIC Purpura fulminans Antiphospholipid syndrome Clots Thrombophilia Thrombus Thrombosis Virchow's triad Trousseau sign of malignancy By site Deep vein thrombosis Bancroft's sign Homans sign Lisker's sign Louvel's sign Lowenberg's sign Peabody's sign Pratt's sign Rose's sign Pulmonary embolism Renal vein thrombosis Bleeding By cause Thrombocytopenia Thrombocytopenic purpura : ITP Evans syndrome TM TTP Upshaw–Schulman syndrome Heparin-induced thrombocytopenia May–Hegglin anomaly Platelet function adhesion Bernard–Soulier syndrome aggregation Glanzmann's thrombasthenia platelet storage pool deficiency Hermansky–Pudlak syndrome Gray platelet syndrome Clotting factor Haemophilia A/VIII B/IX C/XI von Willebrand disease Hypoprothrombinemia/II Factor VII deficiency Factor X deficiency Factor XII deficiency Factor XIII deficiency Dysfibrinogenemia Congenital afibrinogenemia Signs and symptoms Bleeding Bruise Haematoma Petechia Purpura Nonthrombocytopenic purpura By site head Epistaxis Haemoptysis Intracranial haemorrhage Hyphaema Subconjunctival haemorrhage torso Haemothorax Haemopericardium Pulmonary haematoma abdomen Gastrointestinal bleeding Haemobilia Haemoperitoneum Haematocele Haematosalpinx joint Haemarthrosis v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasiaF9, F8, COX8A, AK3, AAVS1, F2, F3, EGF, ALB, F5, SMUG1, CCHCR1, EBP, EIF2AK1, AMT, CCRL2, ST14, TFPI, IFI30, TNFRSF11A, SACM1L, FAM72B, KRT20, DCXR, POLE3, SERHL, AASDHPPT, TP53, RN7SL263P, NBEAL1, RIOX1, HPS6, FAM72A, RIOX2, VIPR1, SMN2, TLR4, FCGRT, CCT, MS4A1, CD38, CFTR, CGA, CTLA4, DBP, EMD, F11, FGG, SPRR2A, G6PD, GAD1, GCY, HLA-A, IL10, KRT31, MNT, TNFRSF11B, SOX3, H3P11

-
Laron Syndrome
Wikipedia
Laron Syndrome - From Man to Mouse: Lessons from Clinical and Experimental Experience . ... "Body composition in untreated adult patients with Laron syndrome (primary GH insensitivity)". Clin. ... "Fifty years on: New lessons from the laron syndrome" . Israel Medical Association Journal . 19 (1): 6–7. ... Retrieved 6 November 2020 . ^ "Laron Syndrome" . GARD: Genetics and Rare Disease Information Center . ... PMID 5916640 . External links [ edit ] Laron+syndrome at the US National Library of Medicine Medical Subject Headings (MeSH) Classification D ICD - 10 : E34.3 ICD - 9-CM : 259.4 OMIM : 262500 245590 MeSH : D046150 DiseasesDB : 7262 External resources eMedicine : ped/1277 Orphanet : 633 v t e Growth and height disorder due to endocrine malfunction Dwarfism Primordial dwarfism Laron syndrome Psychosocial Ateliosis Gigantism v t e Cell surface receptor deficiencies G protein-coupled receptor (including hormone ) Class A TSHR ( Congenital hypothyroidism 1 ) LHCGR ( Luteinizing hormone insensitivity , Leydig cell hypoplasia , Male-limited precocious puberty ) FSHR ( Follicle-stimulating hormone insensitivity , XX gonadal dysgenesis ) GnRHR ( Gonadotropin-releasing hormone insensitivity ) EDNRB ( ABCD syndrome , Waardenburg syndrome 4a , Hirschsprung's disease 2 ) AVPR2 ( Nephrogenic diabetes insipidus 1 ) PTGER2 ( Aspirin-induced asthma ) Class B PTH1R ( Jansen's metaphyseal chondrodysplasia ) Class C CASR ( Familial hypocalciuric hypercalcemia ) Class F FZD4 ( Familial exudative vitreoretinopathy 1 ) Enzyme-linked receptor (including growth factor ) RTK ROR2 ( Robinow syndrome ) FGFR1 ( Pfeiffer syndrome , KAL2 Kallmann syndrome ) FGFR2 ( Apert syndrome , Antley–Bixler syndrome , Pfeiffer syndrome , Crouzon syndrome , Jackson–Weiss syndrome ) FGFR3 ( Achondroplasia , Hypochondroplasia , Thanatophoric dysplasia , Muenke syndrome ) INSR ( Donohue syndrome Rabson–Mendenhall syndrome ) NTRK1 ( Congenital insensitivity to pain with anhidrosis ) KIT ( KIT Piebaldism , Gastrointestinal stromal tumor ) STPK AMHR2 ( Persistent Müllerian duct syndrome II ) TGF beta receptors : Endoglin / Alk-1 / SMAD4 ( Hereditary hemorrhagic telangiectasia ) TGFBR1 / TGFBR2 ( Loeys–Dietz syndrome ) GC GUCY2D ( Leber's congenital amaurosis 1 ) JAK-STAT Type I cytokine receptor : GH ( Laron syndrome ) CSF2RA ( Surfactant metabolism dysfunction 4 ) MPL ( Congenital amegakaryocytic thrombocytopenia ) TNF receptor TNFRSF1A ( TNF receptor associated periodic syndrome ) TNFRSF13B ( Selective immunoglobulin A deficiency 2 ) TNFRSF5 ( Hyper-IgM syndrome type 3 ) TNFRSF13C ( CVID4 ) TNFRSF13B ( CVID2 ) TNFRSF6 ( Autoimmune lymphoproliferative syndrome 1A ) Lipid receptor LRP : LRP2 ( Donnai–Barrow syndrome ) LRP4 ( Cenani–Lenz syndactylism ) LRP5 ( Worth syndrome , Familial exudative vitreoretinopathy 4 , Osteopetrosis 1 ) LDLR ( LDLR Familial hypercholesterolemia ) Other/ungrouped Immunoglobulin superfamily : AGM3, 6 Integrin : LAD1 Glanzmann's thrombasthenia Junctional epidermolysis bullosa with pyloric atresia EDAR ( EDAR hypohidrotic ectodermal dysplasia ) PTCH1 ( Nevoid basal-cell carcinoma syndrome ) BMPR1A ( BMPR1A juvenile polyposis syndrome ) IL2RG ( X-linked severe combined immunodeficiency ) See also cell surface receptorsGHR, GH1, IGF1, IGFBP3, STAT5B, BCL2, SOAT1, SLC35G1, DESI1, TXNIP, TGM2, STAT5A, OPA1, PROP1, CASP3, IGHD, GHRHR, EGF, CASP9, SNORA44

-
Lethal Congenital Contracture Syndrome Type 3
Orphanet
Lethal congenital contracture syndrome type 3 is a rare arthrogryposis syndrome characterized by clinical features identical to Lethal congenital contracture syndrome type 2 (i.e. multiple congenital contactures (typically extended elbows and flexed knees), micrognathia, anterior horn cells degeneration, skeletal muscle atrophy (mainly in the lower limbs), in the absence of hydrops, pterygia or bone fractures), but without bladder enlargement.
-
Cerebro-Facio-Articular Syndrome
Gard
Cerebro-facio-articular syndrome, which is also known as van Maldergem syndrome, is a rare condition that was first described in 1992. Key features of the condition include characteristic facial features, hand abnormalities, moderate to severe intellectual disability, poor muscle tone and joint hyperlaxity . Cerebro-facio-articular syndrome can be caused by changes (mutations) in the DCHS1 or FAT4 genes and is inherited in an autosomal recessive manner.
-
Wrinkly Skin Syndrome
Gard
Wrinkly skin syndrome is a genetic condition characterized by sagging or wrinkly skin, reduced skin elasticity, and delayed closure of the fontanel (a baby's "soft spot" on the top of his/her head). ... It can be caused by mutations in the ATP6VOA2 gene. Wrinkly skin syndrome appears to be represent the mild version of autosomal recessive cutis laxa syndrome type 2 .
-
Wolfram-Like Syndrome
Orphanet
Wolfram-like syndrome is a rare endocrine disease characterized by the triad of adult-onset diabetes mellitus, progressive hearing loss (usually presenting in the first decade of life and principally of low to moderate frequencies), and/or juvenile-onset optic atrophy. Psychiatric (i.e. anxiety, depression, hallucinations) and sleep disorders, the only neurologic abnormalities observed in this disease, have been reported in rare cases. Unlike Wolfram syndrome, patients with Wolfram-like syndrome do not report endocrine or cardiac findings.
-
Oromandibular-Limb Hypogenesis Syndrome
Orphanet
Oromandibular-limb hypogenesis syndromes (OLHS) are a group of dysmorphic complexes (including Charlie M syndrome, Hanhart syndrome and glossopalatine ankylosis; see these terms) characterized by the association of severe asymmetric limb defects (primarily involving distal segments) and abnormalities of the oral cavity and mandible (hypoglossia, aglossia, micrognathia, glossopalatine ankylosis, cleft palate, and gingival anomalies).
-
Charlie M Syndrome
Orphanet
Charlie M syndrome is a rare bone developmental disorder which belongs to a group of oromandibular limb hypogenesis syndromes that includes hypoglossia-hypodactyly and glossopalatine ankylosis (see these terms). The major anomalies which occur commonly in this group are hypoplasia of the mandible, syndactyly and ectrodactyly, small mouth, cleft palate, hypodontia, and facial paralysis. Patients with Charlie M syndrome also present with hypertelorism, absent or conically crowned incisors, and variable degrees of hypodactyly of the hands and feet.
-
Brachytelephalangy-Dysmorphism-Kallmann Syndrome
Orphanet
Brachytelephalangy - dysmorphism - Kallmann syndrome is a developmental anomaly characterized by brachytelephalangy, distinct craniofacial features (prominent square forehead, telecanthus, small nose, malar hypoplasia, smooth philtrum and thin upper lip), and relative to other family members, a short stature. These features may be associated with anosmia and hypogonadotropic hypogonadism (considered as Kallman syndrome ; see this term). Brachytelephalangy - dysmorphism - Kallmann syndrome has been described in a mother and her son and there have been no further descriptions in the literature since 1986.
-
Brooke–fordyce Syndrome
Wikipedia
Brooke–Fordyce syndrome Specialty Dermatology/oncology Brooke–Fordyce syndrome is a condition characterized by multiple trichoepitheliomas . See also [ edit ] List of cutaneous neoplasms associated with systemic syndromes List of cutaneous conditions References [ edit ] Bolognia, Jean L.; et al. (2007).






