-
Median Arcuate Ligament Syndrome
Wikipedia
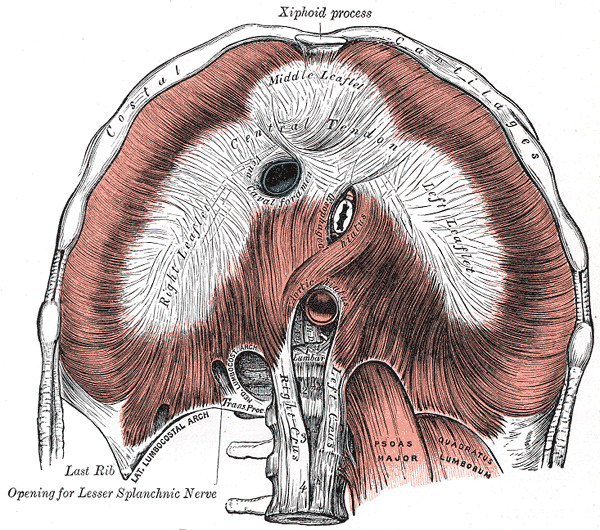
Median arcuate ligament syndrome Other names Celiac artery compression syndrome Celiac axis syndrome Celiac trunk compression syndrome Dunbar syndrome Median arcuate ligament syndrome results from compression of the celiac artery by the median arcuate ligament . ... Specialty Gastroenterology , Vascular Surgery Symptoms Epigastric pain , anorexia , Weight loss Complications Gastroparesis Aneurysm of the superior and inferior pancreaticoduodenal arteries Usual onset 20 to 40 years of age Causes Compression of the celiac artery from the median arcuate ligament Risk factors Female gender Treatment Surgery In medicine , the median arcuate ligament syndrome ( MALS , also known as celiac artery compression syndrome , celiac axis syndrome , celiac trunk compression syndrome or Dunbar syndrome ) is a rare [1] condition characterized by abdominal pain attributed to compression of the celiac artery and the celiac ganglia by the median arcuate ligament . [2] The abdominal pain may be related to meals, may be accompanied by weight loss, and may be associated with an abdominal bruit heard by a clinician. ... David Dunbar and Samuel Marable in 1965. [13] It has also been called Harjola-Marable syndrome and Marable syndrome . [11] See also [ edit ] Nutcracker syndrome Superior mesenteric artery syndrome References [ edit ] ^ "Rare Disease Database: Median Arcuate Ligament Syndrome" . rarediseases.org . ... "US case of the day. Median arcuate ligament syndrome (celiac artery compression syndrome)" . ... Laparoscopic management of median arcuate ligament syndrome". Surg Endosc . 19 (5): 729. doi : 10.1007/s00464-004-6010-x .
-
Tibial Aplasia-Ectrodactyly Syndrome
Orphanet
Tibial aplasia-ectrodactyly syndrome is a rare condition characterized by congenital ectrodactylous limb malformations associated with tibial aplasia or hypoplasia. ... Etiology Two susceptibility loci at 1q42.2-q43 and 6q14.1 have been identified, leading to the hypothesis that this syndrome fits the model of digenic inheritance. Differential diagnosis Overlap with the Gollop-Wolfgang syndrome (see this term) has been described. Genetic counseling The syndrome is generally inherited in an autosomal dominant manner with reduced penetrance.
-
Fg Syndrome 5
Omim
For a general phenotypic description and a discussion of genetic heterogeneity of FG syndrome, see FGS1 (305450). Clinical Features Jehee et al. (2005) reported a Brazilian boy with FG syndrome born to a young and nonconsanguineous couple. ... Mapping In a Brazilian boy with FG syndrome, Jehee et al. (2005) detected an inherited duplication at Xq22.3 by comparative genomic hybridization microarray; the patient's mother was found to be heterozygous for the duplication. Jehee et al. (2005) stated that the duplication maps outside the 4 known loci for FG syndrome, and designated this locus FGS5.
-
Cataract-Ataxia-Deafness-Retardation Syndrome
Omim
Begeer et al. (1991) concluded that this syndrome is distinct from the ataxia-deafness-retardation syndrome (ADR syndrome; 208850) because of the presence of congenital cataract and the later onset of hearing loss and ataxia, which start in infancy in the ADR syndrome.
-
Pagod Syndrome
Orphanet
PAGOD syndrome is a severe developmental syndrome characterized by multiple congenital anomalies including cardiovascular defects, pulmonary hypoplasia, diaphragmatic defects and genital anomalies. ... Clinical description Neonates with PAGOD syndrome present with several visceral anomalies: hypoplasia of right or left lung, diaphragmatic hernia, omphalocele, various cardiac anomalies including, amongst others, atrial septal defect, left ventricular hypoplasia or ventricular septal defect, and great vessels anomalies such as aortic hypoplasia and pulmonary artery hypoplasia or atresia. ... Etiology Etiology is unknown but vitamin A deficiency has been suggested to play a role in the development of the syndrome. Genetic counseling Almost all cases are sporadic, except for 2 siblings, suggesting autosomal recessive inheritance.
-
Intracranial Hypertension Syndrome
Wikipedia
Intracranial hypertension syndrome Specialty Neurology Intracranial hypertension syndrome is characterized by an elevated intracranial pressure , papilledema , and headache with occasional abducens nerve paresis , absence of a space-occupying lesion or ventricular enlargement, and normal cerebrospinal fluid chemical and hematological constituents. [1] [2] References [ edit ] ^ Xue Z, Wang X, Liu F, Hu S, Zhu S, Zhang S, Bu B (February 2009). "Intracranial hypertension syndrome in systemic lupus erythematosus: Clinical analysis and review of the literature". ... "Otologic manifestations of benign intracranial hypertension syndrome: diagnosis and management". Laryngoscope . 97 (8 Pt 2 Suppl 42): 1–17. doi : 10.1288/00005537-198708001-00001 .
-
Valentino's Syndrome
Wikipedia
Cause [ edit ] The cause for Valentino’s syndrome is due to a perforated ulcer located in the duodenum. ... ISBN 9780323416801 . ^ a b c d e Amann CJ, Austin AL, Rudinsky SL (March 2017). "Valentino's Syndrome: A Life-Threatening Mimic of Acute Appendicitis" . ... ISBN 9781481650380 . ^ a b c d Chávez AM, Vázquez AA, López JM, Pavón NL, Gómez JL, Hernández DA, Durón MV (2017-04-22). "Valentino's syndrome: the simulation of an appendicitis" . ... ISBN 9781444165029 . ^ a b c d Sharma R. "Valentino syndrome | Radiology Reference Article" . ... "First Report of Preoperative Imaging Diagnosis of a Surgically Confirmed Case of Valentino's Syndrome" . Journal of Clinical Imaging Science . 4 : 28. doi : 10.4103/2156-7514.133263 .
-
Brachydactyly
Wikipedia
It most often occurs as an isolated dysmelia , but can also occur with other anomalies as part of many congenital syndromes . Brachydactyly can also be a signal that one will be at risk for heart problems as they age. ... Type A6, BDA6 112910 Brachydactyly type A6 or Osebold-Remondini syndrome. Type A7, BDA7 Brachydactyly type A7 or Brachydactyly Smorgasbord type. [1] Type B, BDB (or BDB1) 113000 ROR2 9q22 Brachydactyly type B. ... Type B and E 112440 ROR2 HOXD13 9q22, 2q31-q32 Brachydactyly types B and E combined, Ballard syndrome or Pitt-Williams brachydactyly. Type A1B, BDA1B 607004 5p13.3-p13.2 Brachydactyly type A1, B. Other syndromes [ edit ] In the above brachydactyly syndromes, short digits are the most prominent of the anomalies, but in many other syndromes ( Down syndrome , Rubinstein-Taybi syndrome , etc.), brachydactyly is a minor feature compared to the other anomalies or problems comprising the syndrome. ... Cite journal requires |journal= ( help ) External links [ edit ] Classification D ICD - 10 : Q68.1 ICD - 9-CM : 755.2 - 755.4 MeSH : D059327 DiseasesDB : 29782 SNOMED CT : 43476002 Type A2 [ permanent dead link ] Brachydactyly type A1 at NIH 's Office of Rare Diseases Brachydactyly type A2 at NIH 's Office of Rare Diseases Brachydactyly type A3 at NIH 's Office of Rare Diseases Brachydactyly type A6 at NIH 's Office of Rare Diseases Brachydactyly type A7 at NIH 's Office of Rare Diseases Brachydactyly type B at NIH 's Office of Rare Diseases Brachydactyly type C at NIH 's Office of Rare Diseases Brachydactyly type E at NIH 's Office of Rare Diseases Brachydactyly types B and E combined at NIH 's Office of Rare Diseases v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatumGNAS, HDAC4, SMAD4, TCTEX1D2, PTHLH, ROR2, HOXD13, FBN1, COL2A1, TRPS1, BMPR1B, GDF5, IHH, CHSY1, PDE3A, PRMT7, HOXA13, LMNA, PDE4D, NOG, PRKAR1A, PTCH1, PTH1R, TWIST1, FGFR3, ACAN, RUNX2, B3GLCT, GJB4, CCDC22, IFT172, TCTN3, SIN3A, POC1A, NIPBL, PIGN, KAT6B, ZFPM2, EXOSC2, ADNP, SIK3, SPECC1L, SLC35D1, AFF4, DCPS, TMEM216, KIF7, FAM149B1, IFT81, ANKRD11, SH3PXD2B, DONSON, EOGT, SCAPER, CHST11, KCNK4, TBX22, MRPS16, IFT52, ZDHHC9, PLXND1, POGZ, SPART, IFT140, WASHC5, KIAA0753, PTDSS1, KLLN, KIAA0586, RNU4ATAC, SEMA3E, IQSEC2, MACROH2A1, CHST3, EIF2AK3, ECEL1, CRIPT, LONP1, MAFB, MED12, HUWE1, BHLHA9, CWC27, MIR17HG, CITED2, SEC23B, DEAF1, STAMBP, RAI1, SRCAP, PNPLA6, POP1, MRAS, MYMK, CILK1, RLIM, ZDHHC24, TMEM256, NXN, EVC2, FTO, CPLANE1, UPF3B, PORCN, SIL1, PRDM16, CANT1, FAM111A, TRPV4, NLRC4, WDR19, DOCK6, IFT80, CCDC28B, DYNC2H1, TTC21B, EHMT1, BBS5, CSPP1, ADAMTS10, ARL6, COL25A1, WDR34, TMEM67, BBIP1, PIGS, IFT43, C12orf57, NLRP3, ANTXR2, WDR35, ARHGAP31, TBC1D24, CEP120, POMP, NBAS, DYNC2LI1, SUFU, ARID2, RAB23, INPP5K, WNT4, RIN2, ESCO2, MAGEL2, DLL4, LZTFL1, DYM, NSUN2, SNX14, IMPAD1, IFT57, WDR60, YY1AP1, SLC35C1, CHD7, IARS2, A2ML1, GATA5, IFT122, HDAC8, KIF15, NKX2-6, SLURP1, TRIP11, PTCH2, SMC3, GATA6, GJB3, GJB2, GJA5, GJA1, GHR, GDF1, GATA4, GPX4, GABRD, FZD2, KDSR, FLNB, FLNA, FLII, GLI3, RBPJ, FGFR1, LTBP3, NRAS, NPR2, NOTCH2, NOTCH1, NEK1, MAF, LTBP2, INPPL1, LIG4, LBR, KRAS, KCNJ2, ITGB6, INSR, FGFR2, FGF9, PAX3, BBS1, CHN1, CAMK2G, BRAF, BMP4, BGN, BBS4, ATRX, COL11A1, ATP7A, ATP6V1B2, RERE, ALX3, AKT1, AHSG, COL10A1, COL11A2, FGD1, EFNB1, GPC4, EXTL3, EXT1, EVC, ERF, MEGF8, DVL3, COMP, DVL1, SLC26A2, DHCR7, CTSK, NKX2-5, CSNK2A1, PAH, MEFV, PCNT, SDHB, TNNT3, TGFBR2, TCF12, TBX15, TBX1, MAP3K7, SOS2, SOS1, SMARCD2, SMARCA2, PCYT1A, SLC2A1, SKI, SHOX, SDHD, TRIO, TULP1, WNT5A, ARID1A, PAPSS2, MBTPS1, JAG1, KCNAB2, PPM1D, OFD1, SMC1A, ZIC1, KDM5C, LZTR1, MKKS, ALX1, SHOC2, KAT6A, SDHC, TMEM256-PLSCR3, RRAS, REV3L, PTPN11, MAP2K1, RPS6KA3, RAF1, RASA2, PPP3CA, RB1, DPF2, PPP2R1A, PPP1CB, PITX1, PTEN, RMRP, PIK3R1, PIK3CA, PDE6D, RPL10, RIT1, PDGFRB, PGM3, PTH, TBCEL, GHRH, FZD1, BDA1B, ASAH1, ADAMTS17, PHPT1, KCNB2, CRYGD, COL1A2, QRSL1, FOXC1, BRD2, PADI4, ACSL3, FRAS1, ADAMTS12, RTTN, SOX3, PARK7, SYK, PRPF31, GPC1, SACS, HLA-DRB1, HNRNPR, TBL1XR1

-
Neuroleptic Malignant Syndrome
Wikipedia
"Antipsychotiques atypiques et syndrome malin des neuroleptiques : brève revue de la littérature" [Neuroleptic malignant syndrome and atypical antipsychotics: A brief review] (PDF) . ... "Is risk of neuroleptic malignant syndrome increased in the postpartum period?". ... "Catatonia and neuroleptic malignant syndrome: psychopathology and pathophysiology". ... S2CID 25728796 . ^ Christensen V, Glenthøj B (2001). "[Malignant neuroleptic syndrome or serotonergic syndrome]". Ugeskrift for Lægerer . 163 (3): 301–2. ... PMID 9735957 . ^ Hernández JL, Palacios-Araus L, Echevarría S, Herrán A, Campo JF, Riancho JA (1997). "Neuroleptic malignant syndrome in the acquired immunodeficiency syndrome" .CYP2D6, DRD2, RYR1, ECT, CPT2, GCH1, HTR1A, HTR2A, PIK3C2A, RENBP, TAF1, DPM2, KCNE3, LRRK2, ANKK1, LOC107987479

-
Nephritic Syndrome
Wikipedia
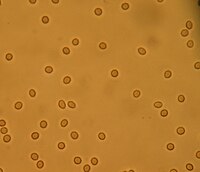
Not to be confused with Nephrotic syndrome . Nephritic syndrome Other names Acute nephritic syndrome [1] Hematuria (one of the symptoms of Nephritic syndrome ) Specialty Nephrology Symptoms Oliguria [2] Causes Infectious, autoimmune, or thrombotic [3] Diagnostic method Urinalysis, kidney biopsy [4] Treatment Antihypertensives [5] Anatomical Kidney with graphical representation of position Nephritic syndrome is a syndrome comprising signs of nephritis , which is kidney disease involving inflammation . ... If the underlying cause is not determined and treated appropriately, it increases the risk of a recurrence of nephritic syndrome or chronic kidney disease (CKD) in the future. [4] Prognosis [ edit ] Because nephritic syndrome is a syndrome and not a disease , the prognosis depends on the underlying cause. ... ISBN 9780781796194 . ^ a b c d e "Acute nephritic syndrome: MedlinePlus Medical Encyclopedia" . www.nlm.nih.gov . Retrieved 2015-10-27 . ^ a b "Acute Nephritis; Nephrosis; Nephritic syndrome information. Patient | Patient" . ... OCLC 41258931 . ^ "Acute nephritic syndrome" . MedlinePlus . July 16, 2019 .
-
Urticarial Syndromes
Wikipedia
There are several distinct urticarial syndromes including: [1] Muckle–Wells syndrome Familial Mediterranean fever Systemic capillary leak syndrome See also [ edit ] Physical urticarias List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). ... ISBN 1-4160-2999-0 . v t e Urticaria and erythema Urticaria ( acute / chronic ) Allergic urticaria Urticarial allergic eruption Physical urticaria Cold urticaria Familial Primary cold contact urticaria Secondary cold contact urticaria Reflex cold urticaria Heat urticaria Localized heat contact urticaria Solar urticaria Dermatographic urticaria Vibratory angioedema Pressure urticaria Cholinergic urticaria Aquagenic urticaria Other urticaria Acquired C1 esterase inhibitor deficiency Adrenergic urticaria Exercise urticaria Galvanic urticaria Schnitzler syndrome Urticaria-like follicular mucinosis Angioedema Episodic angioedema with eosinophilia Hereditary angioedema Erythema Erythema multiforme / drug eruption Erythema multiforme minor Erythema multiforme major Stevens–Johnson syndrome , Toxic epidermal necrolysis panniculitis ( Erythema nodosum ) Acute generalized exanthematous pustulosis Figurate erythema Erythema annulare centrifugum Erythema marginatum Erythema migrans Erythema gyratum repens Other erythema Necrolytic migratory erythema Erythema toxicum Erythroderma Palmar erythema Generalized erythema This dermatology article is a stub .
-
Cortes Lacassie Syndrome
Wikipedia
Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( April 2017 ) Cortes Lacassie syndrome is a rare disease that is characterized by seizures, abnormalities in nails, hair and teeth, and malformed hands and feet. It is named after Fanny Cortes and Yves Lacassie, two researchers from the University of Chile who discovered the disease in 1986. [1] Cortes Lacassie syndrome is considered a rare disease and has only been recorded in one case, leading to death at 31 months. [2] Contents 1 Symptoms 1.1 Related Diseases 2 Cause 3 References Symptoms [ edit ] This disease is characterized by agenesis of fingers and toes, and hypotrophy of fingernails at birth. ... The baby developed acute epileptic seizures , acute renal failure, and hepatorenal syndrome , passing away at 31 months. [1] Related Diseases [ edit ] Cortes Lacassie syndrome is a form of ectodermal dysplasia , postulated to be a new type of the group of tricho-odonto-onychic disorders. ... PMID 3777024 . ^ "Cortes Lacassie syndrome — CheckOrphan" . www.checkorphan.org .
-
Spastic Paraplegia And Evans Syndrome
Omim
Heterogeneity is indicated both by mode of inheritance and by clinical distinctions. Evans syndrome is the simultaneous or sequential occurrence of Coombs-positive hemolytic anemia and immune thrombocytopenia without a known underlying etiology (Evans and Duane, 1949). ... Ahmed et al. (1996) described 2 Saudi brothers aged 13 and 9 years, the offspring of first-cousin parents, who walked on their toes from an early age and were initially diagnosed as congenital spastic diplegia. Evidence of the Evans syndrome began at age 5 in the older brother and age 2.5 in the younger brother. Rapid deterioration of functional motor ability followed the development of Evans syndrome. It is noteworthy that in the family a sister had died at one year of age with intracranial hemorrhage; she was said to have had hemolytic anemia and thrombocytopenia. Further details of investigations could not be traced. Thus, 3 sibs with Evans syndrome among the offspring of a consanguineous marriage were observed, suggesting autosomal recessive inheritance. Although genetic predisposition in Evans syndrome has perhaps not been specifically noted, familial clustering of cases of autoimmune hemolytic anemia and autoimmune thrombocytopenia as individual entities is well known.
-
Runting-Stunting Syndrome In Broilers
Wikipedia
Please help improve it to make it understandable to non-experts , without removing the technical details. ( July 2018 ) ( Learn how and when to remove this template message ) Runting-stunting syndrome in broilers is a syndrome described in broilers since the 1940s, but often with specific etiological appellations ( viral enteritis , malabsorption syndrome , brittle bone disease , infectious proventriculitis , helicopter disease and pale bird syndrome ). ... References [ edit ] ^ http://www.thepoultrysite.com/articles/1110/runtingstunting-syndrome-in-broilers
-
Otopalatodigital Syndrome Spectrum Disorder
Orphanet
Otopalatodigital syndrome spectrum disorder is a primary bone dysplasia and encompasses a group of congenital anomalies that are characterized by skeletal dysplasia of varying clinical severity and an X linked dominant pattern of inheritance. This group includes otopalatodigital syndrome type 1 and 2 (OPD1, OPD2) which are characterized in affected males by cleft palate, conductive hearing loss, craniofacial abnormalities and skeletal dysplasia; Melnick-Needles syndrome (MNS) which displays skeletal deformities in females and embryonic or perinatal lethality in most males; frontometaphyseal dysplasia (FMD); and terminal osseous dysplasia - pigmentary defects.
-
Kousseff Syndrome
Orphanet
Kousseff syndrome is characterized by the association of conotruncal heart defects, myelomeningocele and craniofacial dysmorphism similar to that seen in monosomy 22q11 (see this term). ... Etiology The identification, by FISH, of 22q11.2 deletions in the majority of reported cases (including the original cases described by Kousseff) indicated that this syndrome is part of the variable clinical spectrum of monosomy 22q11. However, the absence of a 22q11.2 deletion in one patient presenting with myelomeningocele, tetralogy of Fallot, microcephaly, hydrocephalus, hypoplasia of the corpus callosum, and moderate developmental delay leaves open the possibility that Kousseff syndrome is a distinct genetic entity.
-
German Syndrome
Orphanet
German syndrome is an autosomal recessive arthrogryposis syndrome, described in 5 cases. Three of the four known families with affected children were Ashkenazi Jews. German syndrome is characterized by arthrogryposis, hypotonia-hypokinesia sequence, and lymphedema.
-
Early-Onset Epileptic Encephalopathy-Cortical Blindness-Intellectual Disability-Facial Dysmorphism Syndrome
Orphanet
Early-onset epileptic encephalopathy-cortical blindness-intellectual disability-facial dysmorphism syndrome is a rare, syndromic intellectual disability syndrome characterized by cortical blindness, different types of seizures, intellectual disability with limited or absent speech, and dysmorphic facial features.
-
Westerhof Syndrome
Wikipedia
Westerhof syndrome Specialty Dermatology Westerhof syndrome is a cutaneous condition inherited in an autosomal dominant fashion, characterized by congenital hypopigmented macules . [1] See also [ edit ] Watson syndrome List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).
-
Retinal Ischemic Syndrome-Digestive Tract Small Vessel Hyalinosis-Diffuse Cerebral Calcifications Syndrome
Orphanet
A rare systemic disease characterized by progressive hyalinosis involving capillaries, arterioles and small veins of the digestive tract, kidneys, and retina, associated with idiopathic cerebral calcifications, manifesting with severe diarrhea (with rectal bleeding and malabsorption), nephropathy (with renal failure and systemic hypertension), chorioretinal scarring, and subarachnoid hemorrhage. Poikiloderma and premature greying of the hair may be additionally observed.



