A rare multiple congenital anomaly syndrome characterized by a distinct facial phenotype, intellectual disability, epilepsy, Hirschsprung disease (HSCR) and variable congenital malformations. ... It seems probable that Mowat-Wilson syndrome (MWS) is underdiagnosed, particularly in patients without HSCR. ... Differential diagnosis Differential diagnoses include Pitt-Hopkins, Goldberg-Shprintzen megacolon, Smith-Lemli-Opitz and Angelman syndromes. Antenatal diagnosis Prenatal diagnosis is available for subsequent pregnancies of parents with an affected child.
Mowat-Wilson syndrome is a genetic condition that affects many parts of the body. ... Chronic constipation also occurs frequently in people with Mowat-Wilson syndrome who have not been diagnosed with Hirschsprung disease. Other features of Mowat-Wilson syndrome include short stature, seizures, heart defects, and abnormalities of the urinary tract and genitalia. ... Although many different medical issues have been associated with Mowat-Wilson syndrome, not every individual with this condition has all of these features. Frequency The prevalence of Mowat-Wilson syndrome is unknown. More than 200 people with this condition have been reported in the medical literature.
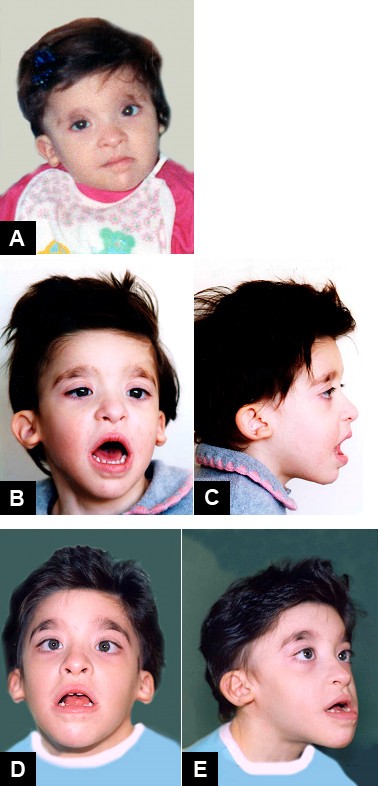
Diagnosis Formal clinical diagnostic criteria for Mowat-Wilson syndrome (MWS) have not been published. ... An individual with Mowat-Wilson syndrome at (a) one month, (b) two months, (c) five years, (d) 13 years, (e) 20 years, and (f) 21 years. ... Differential Diagnosis Many of the congenital anomalies seen in Mowat-Wilson syndrome (MWS) can be seen as isolated anomalies in an otherwise normal individual. ... Angelman syndrome is caused by disruption of maternally imprinted UBE3A located within the 15q11.2-q13 Angelman syndrome / Prader-Willi syndrome (AS/PWS) region. ... Management Clinical management guidelines for Mowat-Wilson syndrome have been published (full text) [Ivanovski et al 2018].
Mowat-Wilson syndrome has many clinical features in common with Goldberg-Shprintzen syndrome (609460) but the 2 disorders are genetically distinct (Mowat et al., 2003). Goldberg-Shprintzen syndrome is caused by mutation in the KIAA1279 gene (609367) located on 10q. ... This patient thus appeared to represent an intermediate stage between the full-blown Hirschsprung disease-mental retardation syndrome and Hirschsprung disease-mental retardation syndrome without Hirschsprung disease. ... One of the patients with a deletion mutation had previously been reported by Tanaka et al. (1993) to have Goldberg-Shprintzen syndrome. Amiel et al. (2001) stated that 2 other patients reported to have Goldberg-Shprintzen syndrome (Hurst et al., 1988, patient 3; Ohnuma et al., 1997) most likely had Mowat-Wilson syndrome. ... Engenheiro et al. (2008) noted that both patients were only properly diagnosed with Mowat-Wilson syndrome after initial cytogenetic findings were investigated further, suggesting that the syndrome may be underdiagnosed.
Congenital disorder in which the pituitary stalk and pituitary are hypoplastic Pituitary stalk interruption syndrome (PSIS) Other names Ectopic neurohypophysis The location of the pituitary gland within the skull (indicated in orange) Specialty Endocrinology , neurology , neonatology , paediatrics Symptoms Hypoglycaemia, jaudice, micropenis, cryptorchidism, etc. ... Due to the before-mentioned factors, mortality and morbidity is higher than that of the general population, particularly during the first 2 years of life. [3] Epidemiology [ edit ] The prevalence of PSIS is unknown, however, some 1,000 cases have been reported either with or without the full triad. [3] References [ edit ] ^ a b c d e "Pituitary stalk interruption syndrome" . Genetic and Rare Diseases Information Center (GARD) – an NCATS Program . ... (November 2015). "Pituitary Stalk Interruption Syndrome from Infancy to Adulthood: Clinical, Hormonal, and Radiological Assessment According to the Initial Presentation" . ... PMID 26562670 . ^ a b c d e f Brauner R. "Pituitary stalk interruption syndrome" . Orphanet . Retrieved 2018-08-11 . External links [ edit ] Classification D External resources GARD : Pituitary stalk interruption syndrome Orphanet : 95496
Pituitary stalk interruption syndrome (PSIS) is a congenital abnormality of the pituitary that is responsible for pituitary deficiency and is usually characterized by the triad of a very thin or interrupted pituitary stalk, an ectopic (or absent) posterior pituitary (EPP) and hypoplasia or aplasia of the anterior pituitary visible on MRI.
Pituitary stalk interruption syndrome (PSIS) is a congenital abnormality of the pituitary gland characterized by the triad of a very thin or interrupted pituitary stalk , a misplaced (ectopic) or absent posterior pituitary and a small or absent anterior pituitary , with permanent growth hormone (GH) deficit.
A rare genetic, syndromic glomerular disorder characterized by the association of nephropathy presenting as persistent proteinuria or overt nephrotic syndrome, Wilms tumor and genitourinary structural defects. ... Clinical description Presentation is typically of infantile nephrotic syndrome progressing to end-stage renal disease (ESRD) within 1 to 15 years. ... Careful renal ultrasonography (US) for WT should be performed in any subject found to have early onset nephrotic syndrome. Diffuse mesangial sclerosis is the classic finding on renal biopsy, although other types of nephropathy are common. ... Differential diagnosis The differential diagnosis includes WAGR syndrome, Frasier syndrome, Meacham syndrome, congenital nephrotic syndrome of the Finnish type, idiopathic nephrotic syndrome, and isolated diffuse mesangial sclerosis Antenatal diagnosis Undervirilized / ambiguous genitalia in 46,XY individuals may sometimes be noted on prenatal US. ... Management and treatment Multidisciplinary management should involve a nephrologist, urologist, endocrinologist, oncologist and surgeons. The nephrotic syndrome is resistant to corticosteroids and immunosuppressive drugs.
Denys-Drash syndrome is a condition that affects the kidneys and genitalia . Denys-Drash syndrome is characterized by kidney disease that begins within the first few months of life. ... Because they do not have all the features of the condition, females are usually given the diagnosis of isolated nephrotic syndrome. Frequency The prevalence of Denys-Drash syndrome is unknown; at least 150 affected individuals have been reported in the scientific literature. ... WT1 gene mutations that cause Denys-Drash syndrome lead to the production of an abnormal protein that cannot bind to DNA. ... Denys-Drash syndrome has features similar to another condition called Frasier syndrome, which is also caused by mutations in the WT1 gene.
See also Meacham syndrome (608978) and Frasier syndrome (136680), allelic disorders with similar clinical features. ... The authors presented a patient without ambiguous genitalia, and suggested that all girls with Wilms tumor should be considered at risk for the Drash syndrome. Moorthy et al. (1987) suggested that some of the patients reported as cases of Drash syndrome in fact had Frasier syndrome (136680). ... Schumacher et al. (1998) identified WT1 mutations in 10 children with early-onset nephrotic syndrome. Two genotypically female girls had isolated congenital/infantile nephrotic syndrome (NPHS4; 256370). ... End-stage renal disease was reached either concomitantly or within 4 months after onset of nephrotic syndrome in 7 patients. Four children developed Wilms tumor either before or concomitant with nephrotic syndrome. ... Mueller (1994) reviewed the Denys-Drash syndrome in detail on the basis of 150 reported cases.
Denys-Drash syndrome is a condition that affects the kidneys and genitalia. ... In addition, up to 90 percent of people with this condition develop a rare form of kidney cancer known as Wilms tumor . Males with Denys-Drash syndrome have gonadal dysgenesis, a condition in which the external genitalia do not look clearly male or clearly female ( ambiguous genitalia ) or the genitalia appear to be completely female. ... For this reason, females with this condition may be diagnosed with isolated nephrotic syndrome . Denys-Drash syndrome is caused by mutations in the WT1 gene.
The differential diagnosis of Silver-Russell syndrome (SRS) includes any condition that can cause intrauterine growth restriction and short stature. ... Uniparental disomy for several chromosomes have been reported to cause an SRS-like phenotype, including chromosomes 6, 14 (Temple syndrome), 16, and 20 [Sachwitz et al 2016, Wakeling et al 2017, Geoffron et al 2018]. ... Table 3 summarizes disorders to consider in the differential diagnosis of Silver-Russell syndrome. Table 3. Other Disorders with IUGR and Poor Postnatal Growth to Consider in the Differential Diagnosis of Silver-Russell Syndrome View in own window Disorder Gene(s) MOI Additional Clinical Features of This Disorder Overlapping w/SRS Distinguishing from SRS Monogenic disorders Three M syndrome CCDC8 CUL7 OBSL1 AR Clinodactyly Frontal bossing Relative macrocephaly Triangular facies Pectus excavatum Rib hypoplasia Short neck IMAGe syndrome CDKN1C AD 1 Frontal bossing Macrocephaly Adrenal hypoplasia Adrenal insufficiency Metaphyseal dysplasia Bloom syndrome BLM AR Triangular facies 5th-finger clinodactly Café au lait spots Abnormal sister chromatid exchange Dolichocephaly Microcephaly Nijmegen breakage syndrome NBN AR Café au lait spots Chr instability Microcephaly Sloping forehead Telangiectasia Warsaw breakage syndrome (OMIM 613398) DDX11 AR 5th-finger clinodactyly Chr instability Deafness Microcephaly Sloping forehead Epicanthal folds Fanconi anemia >20 genes 2 AR AD XL Café au lait spots Chr instability Absent thumb(s) or thumb hypoplasia Microcephaly Radial anomalies ↑ malignancy risk Meier-Gorlin syndrome (OMIM 224690) CDC45 CDC6 CDT1 GMNN MCM5 ORC1 ORC4 ORC6 AR AD Frontal bossing Absent patellae Microcephaly Microtia Small mouth Insulin growth factor 1 resistance (incl deletion 15q26.1) 3 IGF1R AR AD Clinodactyly Dental anomalies Global DD Microcephaly Synophrys Chromosome abnormalities Diploiod/triploid mixoploidy 4 Limb asymmetry Global DD Microcephaly Temple syndrome (maternal UPD14, paternal chr 14 deletion or loss of methylation at 14q32) (OMIM 616222) Many features overlap w/SRS. ... Typically a CDKN1C pathogenic variant causing IMAGe syndrome is inherited in an autosomal dominant manner; however, only maternal transmission of the pathogenic variant results in IMAGe syndrome. 2. ... Males with micropenis and females with internal genitourinary anomalies (some are born with Mayer-Rokitansky-Kuster-Hauser syndrome) benefit from referral to a multidisciplinary disorders of sex development (DSD) center.
Silver-Russell syndrome is characterized by growth retardation with antenatal onset, characteristic facies and limb asymmetry. ... Differential diagnosis Differential diagnosis includes intrauterine growth retardation due to impaired placental function, structural or mosaic chromosomal abnormalities, neonatal progeria (Wiedemann-Rautenstrauch syndrome), 3M syndrome and Mulibrey dwarfism (see these terms).
One such family has been reported. Silver-Russell syndrome (SRS; 180860), which has overlapping features, is caused by epigenetic alteration in the chromosome 11p15 region. ... Molecular Genetics In a 4-generation family with GRDF, in which Silver-Russell syndrome-associated molecular alterations had been excluded, Begemann et al. (2015) performed exome sequencing and identified a heterozygous nonsense mutation in the IGF2 gene (S64X; 147470.0004) that segregated fully with the disorder.
The possibility of an X-linked form was raised by Partington (1985) on the basis of the following observations: 2 brothers, aged 7 and 4, had prenatal growth retardation, triangular facies and cafe-au-lait (CAL) spots. Both had asthma. The mother was 160 cm tall and had CAL spots. Her 4 brothers were tall with no spots. Of her 5 sisters, 3 were over 168 cm tall and had no spots; 2 were 156 cm tall and had CAL spots. Partington (1985) suggested that this may represent X-linked inheritance with severe expression in males and mild expression in females. In the full report with illustrations (Partington, 1986), the pigmentary anomaly was presented as quite different from cafe-au-lait spots.
Opposite epimutations, namely hypermethylation at the same region on 11p15, are observed in about 5 to 10% of patients with Beckwith-Wiedemann syndrome (BWS; 130650), an overgrowth syndrome (Bartholdi et al., 2009). Clinical Features Silver-Russell syndrome (SRS) was reported independently by Silver et al. (1953) and Russell (1954). ... Although each of these authors emphasized different phenotypic features, the whole picture was later identified as the 'Russell-Silver syndrome' (Patton, 1988). Chitayat et al. (1988) described hepatocellular carcinoma in a 4-year-old boy with Russell-Silver syndrome. ... Donnai et al. (1989) described unusually severe Silver-Russell syndrome in 3 children with pre- and postnatal growth deficiency. ... Cytogenetics Chromosome 17 Ramirez-Duenas et al. (1992) observed severe Russell-Silver syndrome in a girl with translocation t(17;20)(q25;q13).
Russell-Silver syndrome is a growth disorder characterized by slow growth before and after birth. ... Frequency The exact incidence of Russell-Silver syndrome is unknown. Worldwide estimates range from 1 in 30,000 to 1 in 100,000 people. ... Abnormalities involving these genes appear to be responsible for many cases of Russell-Silver syndrome. Researchers suspect that 30 to 50 percent of all cases of Russell-Silver syndrome result from changes in a process called methylation on the short (p) arm of chromosome 11 at position 15 (11p15). ... In about 40 percent of people with Russell-Silver syndrome, the cause of the condition is unknown. ... Learn more about the genes and chromosomes associated with Russell-Silver syndrome H19 IGF2 chromosome 11 chromosome 7 Inheritance Pattern Most cases of Russell-Silver syndrome are sporadic, which means they occur in people with no history of the disorder in their family.
Branchio-oto-renal syndrome Other names BOR syndrome , Branchiootorenal syndrome Branchio-oto-renal syndrome has an autosomal dominant pattern of inheritance Specialty Medical genetics Symptoms Ear abnormalities [1] Causes Mutations in genes, EYA1, SIX1, and SIX5 [2] Diagnostic method Laboratory test results, Physical exam [3] Treatment Branchial fistula may need surgery [3] Branchio-oto-renal syndrome (BOR) , is an autosomal dominant genetic disorder involving the kidneys , ears, and neck. ... A 2014 review found 250 such cases in the country of Japan. [9] See also [ edit ] Lachiewicz Sibley syndrome Branchio-oculo-facial syndrome References [ edit ] ^ a b c "Branchio Oto Renal Syndrome - NORD (National Organization for Rare Disorders)" . ... Retrieved 2015-11-29 . ^ a b "Branchiootorenal syndrome" . Genetics Home Reference . 2015-11-23 . ... "A family with the branchio‐oto‐renal syndrome: clinical and genetic correlations". ... External links [ edit ] Classification D ICD - 10 : Q87.0 OMIM : 113650 MeSH : D019280 DiseasesDB : 32599 Scholia has a topic profile for Branchio-oto-renal syndrome . v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
Although Melnick et al. (1978) maintained that the BOR syndrome is distinct from the BO syndrome because in the latter condition renal anomaly is absent and deafness is not a constant feature, Cremers and Fikkers-van Noord (1980) concluded that the 2 syndromes are in fact a single entity. ... Carmi et al. (1983) observed a man with the BOR syndrome and crossed renal ectopia who fathered 3 children born with bilateral renal agenesis and the Potter syndrome. ... Legius et al. (1990) described father and son with a branchial cleft syndrome mixing characteristics of the BOR syndrome with those of the branchiooculofacial syndrome (BOFS; 113620). ... Lin et al. (1992) concluded that the patients of Legius et al. (1990) indeed had the BOF syndrome and that this entity is distinct from the BOR syndrome. ... No mutations were detected in any subjects with atypical BOR syndrome or OFC syndrome. Vervoort et al. (2002) noted that in up to one-half of reported cases of BOR syndrome, EYA1 screening was negative, suggesting genetic heterogeneity.
Branchiootorenal syndrome is characterized by birth defects or anomalies of tissues in the neck, malformations of the external ear, hearing loss, and kidney malformations.
A number sign (#) is used with this entry because of evidence that branchiootorenal syndrome-2 (BOR2) is caused by heterozygous mutation in the SIX5 gene (600963) on chromosome 19q13. For a phenotypic description and a discussion of genetic heterogeneity of the branchiootorenal syndrome, see BOR1 (113650). Clinical Features Hoskins et al. (2007) reported 5 patients with a clinical diagnosis of BOR who carried heterozygous mutations in the SIX5 gene. ... The SIX5 gene was considered a good candidate for BOR owing to its similarity to SIX1 (601205), mutations in which cause branchiootic syndrome-3 (608389), and data on C. elegans on its direct interaction with eya-1 (601653), mutations in which also cause BOR (Li et al., 2004). ... This observation, in addition to the extreme rarity of SIX5 mutations, caused Krug et al. (2011) to reconsider the role of SIX5 in branchiootorenal syndrome etiology.
Branchiootorenal (BOR) syndrome is characterized by branchial arch anomalies (branchial clefts, fistulae, cysts), hearing impairment (malformations of the auricle with pre-auricular pits, conductive or sensorineural hearing impairment), and renal malformations (urinary tree malformation, renal hypoplasia or agenesis, renal dysplasia, renal cysts). ... Antenatal diagnosis Prenatal testing can be proposed to families in which the disease-causing mutation has been identified, but genetic counseling is difficult because of the clinical heterogeneity between individuals. Genetic counseling BOR syndrome is transmitted in an autosomal dominant manner.
Molecular Genetic Testing Used in Rubinstein-Taybi Syndrome View in own window Gene 1, 2 Proportion of Rubinstein-Taybi Syndrome Attributed to Pathogenic Variants in Gene Proportion of Probands with a Pathogenic Variant 3 Detectable by Method Sequence analysis 4 Gene-targeted deletion/duplication analysis 5 CREBBP 50%-60% 6 ~80% ~20% 7 EP300 8%-10% 8 >99% <1% 9 Unknown 10 ~30% NA 1. ... Broad/angulated thumbs and halluces may be seen in the FGFR -related craniosynostosis syndromes (e.g., Pfeiffer syndrome, Apert syndrome), in Saethre-Chotzen syndrome, and in Greig cephalopolysyndactyly syndrome. ... Other Genes of Interest in the Differential Diagnosis of Rubinstein-Taybi Syndrome (RSTS) View in own window Gene(s) DiffDx Disorder MOI Clinical Features of DiffDx Disorder Overlapping w/RSTS Distinguishing from RSTS FGFR1 FGFR2 Pfeiffer syndrome & Apert syndrome (see FGFR -related craniosynostosis syndromes) AD Broad/angulated thumbs & halluces Bicoronal craniosynostosis or cloverleaf skull Distinctive facial features TWIST1 Classic Saethre-Chotzen syndrome AD Broad/angulated thumbs & halluces Coronal synostosis (unilateral or bilateral), facial asymmetry, ptosis, & characteristic appearance of the ear (small pinna w/prominent crus) Syndactyly of digits 2 & 3 of the hand variably present Mild-to-moderate DD & ID reported; normal intelligence is more common. ... Floating-Harbor syndrome is caused by a pathogenic variant in SRCAP , which encodes an SNF2-related chromatin-remodeling factor that serves as a coactivator for CREB-binding protein. ... The genetic basis of Keipert syndrome is unknown (OMIM 255980). Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with Rubinstein-Taybi syndrome (RSTS), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended [Wiley et al 2003].
A number sign (#) is used with this entry because the phenotype is a contiguous gene syndrome caused by deletion of chromosome 16p13.3, affecting the CREBBP (600140), DNASE1 (125505), and TRAP1 (HSP75; 606219) genes. Rubinstein-Taybi syndrome-1 (RSTS1; 180849) is caused by point mutations or deletions within the CREBBP gene. ... Bartsch et al. (1999) suggested that patients with severe RSTS and large deletions had a distinct contiguous gene syndrome. Bartsch et al. (2006) provided further analysis of the 2 patients with severe RSTS reported by Bartsch et al. (1999) and a third patient reported by Bartsch et al. (2002). ... Bartsch et al. (2006) noted that Kimura et al. (1993) had described a Japanese boy with RSTS and features of DiGeorge syndrome (188400) who died at age 20 months. ... Bartsch et al. (2006) suggested that 'severe RSTS' is distinct from RSTS and represents a contiguous gene syndrome. The authors suggested that the size of the deletion interval should be determined in such patients because of the risk of critical infections and high mortality.
Genetic Heterogeneity of Rubinstein-Taybi Syndrome Rubinstein-Taybi syndrome-1 (RSTS1) constitutes about 50 to 70% of patients with the disorder. ... See also chromosome 16p13.3 deletion syndrome (610543), a severe form of Rubinstein-Taybi syndrome resulting from a contiguous gene deletion involving the CREBBP gene as well as other neighboring genes. ... Incomplete Rubinstein-Taybi Syndrome Cotsirilos et al. (1987) described 2 sibs and their mother with a syndrome that they reported as similar to Rubinstein-Taybi syndrome. ... Hendrich and Bickmore (2001) reviewed human disorders which share in common defects of chromatin structure or modification, including the ATR-X spectrum of disorders (301040), ICF syndrome (242860), Rett syndrome (312750), Rubinstein-Taybi syndrome, and Coffin-Lowry syndrome (303600). ... Beets et al. (2014) stated that the birth prevalence of Rubinstein-Taybi syndrome is 1 in 100,000-125,000. Nomenclature Although the acronym RTS is sometimes used for Rubinstein-Taybi syndrome, the use of this acronym for 2 other syndromes, Rothmund-Thomson syndrome (268400) and Rett syndrome, may lead to confusion; hence, use of the symbol RSTS is recommended.
Other than previously thought, persons with Rubinstein-Taybi syndrome under the age of 40 have no increased risk of getting malignant tumors. It is unclear whether this risk is increased in the elderly with Rubinstein-Taybi syndrome. Etiology Causes of Rubinstein-Taybi syndrome include: a microdeletion of chromosome 16p13.3 or chromosome 22q13.2, a variant in CREB-binding protein ( CREBBP , 16p13.3) or a variant in E1A-binding protein p300 ( EP300 , 22q13.2). ... A cytogenetic or molecular abnormality can be detected in about 65% of individuals. Differential diagnosis The syndrome can sometimes be difficult to differentiate from Saethre-Chotzen syndrome, Floating Harbor syndrome and Cornelia de Lange syndrome. ... Prenatal ultrasound only rarely allows a reliable diagnosis. Genetic counseling The syndrome is almost always sporadic, most cases resulting from de novo mutations. ... If a person with Rubinstein-Taybi syndrome is able to reproduce, the recurrence risk is 50% as the transmission is autosomal dominant.
A number sign (#) is used with this entry because Rubinstein-Taybi syndrome-2 (RSTS2) is caused by heterozygous mutation in the EP300 gene (602700) on chromosome 22q13. Most, if not all, mutations occur de novo. Description Rubinstein-Taybi syndrome (RSTS) is a multiple congenital anomaly syndrome characterized by mental retardation, postnatal growth deficiency, microcephaly, broad thumbs and halluces, and dysmorphic facial features. ... For a discussion of genetic heterogeneity of Rubinstein-Taybi syndrome, see RSTS1 (180849). Clinical Features Roelfsema et al. (2005) reported 3 unrelated patients with RSTS2. ... Woods et al. (2014) reported a 5-year-old Caucasian male with a phenotype suggesting Cornelia de Lange syndrome (CDLS; 122470) in whom no mutations were found in CDLS-related genes. ... Three patients had overlapping features with Floating-Harbor syndrome (136140), including thin upper vermilion, long nose, and low-hanging columella in 2 and delayed bone age in 2.
Rubinstein-Taybi syndrome (RTS) is a syndrome characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability. The syndrome may be caused by a mutation in the CREBBP or EP300 gene, or as the result of a very small loss (microdeletion) of genetic material from the short (p) arm of chromosome 16 .
Orofaciodigital syndromes refers to numerous conditions in which the oral cavity (mouth, tongue, teeth, and jaw), facial structures (head, eyes, and nose), and digits (fingers and toes) may be formed differently. When changes happen to many different parts of the body, this is called a syndrome. The literature reports up to thirteen types of orofaciodigital syndrome, but research is necessary to confirm and clarify all of these types. ... See below for a list of orofaciodigital syndromes. These types are defined by certain symptoms or characteristics in addition to the those affecting the oral cavity, facial structures, and digits. Click on the embedded links to learn more about each type: Orofaciodigital syndrome 1 Orofaciodigital syndrome 8 Orofaciodigital syndrome 2 Orofaciodigital syndrome 9 Orofaciodigital syndrome 3 Orofaciodigital syndrome 10 Orofaciodigital syndrome 4 Orofaciodigital syndrome 11 Orofaciodigital syndrome 5 Orofaciodigital syndrome 12 Orofaciodigital syndrome 6 Orofaciodigital syndrome 13 Orofaciodigital syndrome 7* *now considered identical to type 1
Oral-facial-digital syndrome Other names Orofaciodigital syndrome Specialty Rheumatology , medical genetics Oral-facial-digital syndrome is a group of at least 13 related conditions that affect the development of the mouth , facial features , and digits in between 1 in 50,000 to 250,000 newborns with the majority of cases being type I ( Papillon-League-Psaume syndrome ). [1] Contents 1 Type 2 References 3 External links Type [ edit ] The different types are:s [2] Type I, Papillon-League-Psaume syndrome Type II, Mohr syndrome [3] Type III, Sugarman syndrome Type IV, Baraitser-Burn syndrome [4] Type V, Thurston syndrome [5] Type VI, Varadi-Papp syndrome [6] Type VII, Whelan syndrome [7] Type VIII, Oral-facial-digital syndrome, Edwards type [8] (not to be confused with Edwards syndrome ) Type IX, OFD syndrome with retinal abnormalities [9] Type X, OFD with fibular aplasia [10] Type XI, Gabrielli syndrome [11] References [ edit ] ^ "Oral-facial-digital syndrome - Genetics Home Reference" . ^ "National Organization for Rare Disorders" . ... Retrieved 2015-03-02 . ^ Online Mendelian Inheritance in Man (OMIM): MOHR SYNDROME - 252100 ^ Online Mendelian Inheritance in Man (OMIM): OROFACIODIGITAL SYNDROME IV; OFD4 - 258860 ^ Online Mendelian Inheritance in Man (OMIM): OROFACIODIGITAL SYNDROME V; OFD5 - 174300 ^ Online Mendelian Inheritance in Man (OMIM): OROFACIODIGITAL SYNDROME VI; OFD6 - 277170 ^ Online Mendelian Inheritance in Man (OMIM): OROFACIODIGITAL SYNDROME VII; OFD7 - 608518 ^ Online Mendelian Inheritance in Man (OMIM): OROFACIODIGITAL SYNDROME VIII; OFD8 - 300484 ^ Online Mendelian Inheritance in Man (OMIM): OROFACIODIGITAL SYNDROME IX; OFD9 - 258865 ^ Online Mendelian Inheritance in Man (OMIM): OROFACIODIGITAL SYNDROME X; OFD10 - 165590 ^ Online Mendelian Inheritance in Man (OMIM): OROFACIODIGITAL SYNDROME XI; OFD11 - 612913 External links [ edit ] Classification D ICD - 10 : Q87.0 MeSH : D009958 External resources Orphanet : 140997 This genetic disorder article is a stub .
X-linked adrenal hypoplasia congenita is a disorder that mainly affects males. It involves many hormone-producing (endocrine) tissues in the body, particularly a pair of small glands on top of each kidney called the adrenal glands. These glands produce a variety of hormones that regulate many essential functions in the body. One of the main signs of this disorder is adrenal insufficiency, which occurs when the adrenal glands do not produce enough hormones. Adrenal insufficiency typically begins in infancy or childhood and can cause vomiting, difficulty with feeding, dehydration, extremely low blood sugar (hypoglycemia), and shock.
Mackay et al. (2006) reported 2 unrelated TNDM patients who had loss of maternal methylation both at 6q24 and at the centromeric DMR on 11p15.5 (KCNQ1OT1; 604115), which is involved in imprinting abnormalities in Beckwith-Wiedemann syndrome. Both patients presented with intrauterine growth retardation and TNDM without features of overgrowth. ... They reported the fifth case of paternal uniparental disomy of chromosome 6 associated with classic transient neonatal diabetes mellitus and estimated that uniparental disomy of chromosome 6 accounts for approximately one-fifth of cases of this syndrome. Since either duplication of a portion of chromosome 6q or uniparental disomy have been associated with transient neonatal diabetes mellitus, overexpression of an imprinted gene in this disorder is suggested. ... This observation demonstrated that mitotic recombination of chromosome 6 can also give rise to uniparental disomy and neonatal diabetes, a situation similar to that observed in Beckwith-Wiedemann syndrome (BWS; 130650), another imprinting disorder. ... Mackay et al. (2006) proposed the existence of a maternal hypomethylation syndrome and suggested that any patient with methylation loss at 1 maternally-methylated locus might also manifest methylation loss at other loci, potentially complicating or even confounding the clinical presentation.
A number sign (#) is used with this entry because this form of transient neonatal diabetes mellitus is caused by mutation in the ABCC8 gene (600509). For a phenotypic description and a discussion of genetic heterogeneity of transient neonatal diabetes mellitus, see 601410. From a group of 73 patients with neonatal diabetes, Babenko et al. (2006) screened the ABCC8 gene in 34 who did not have alterations in chromosome 6q (see 601410) or mutations in the KCNJ11 (600937) or GCK (138079) genes. They identified heterozygosity for 5 different mutations (see, e.g., 600509.0019 and 600509.0020) in 7 patients with transient neonatal diabetes mellitus. They also identified heterozygosity for mutations (600509.0017 and 600509.0018) in 2 patients with permanent neonatal diabetes (606176).
A number sign (#) is used with this entry because of evidence that transient neonatal diabetes mellitus-3 (TNDM3) is caused by heterozygous mutation in the KCNJ11 gene (600937) on chromosome 11p15. For a phenotypic description and a discussion of genetic heterogeneity of transient neonatal diabetes mellitus, see 601410. Clinical Features Yorifuji et al. (2005) studied members of a 4-generation family with dominantly inherited diabetes mellitus observed in 3 generations. None of the patients in this family had permanent neonatal diabetes. The proband had transient neonatal diabetes, and his paternal grandfather had been diagnosed with diabetes at 3 years of age. The proband's paternal aunt at age 26 developed gestational diabetes, which was transient, but was diagnosed with adult-onset diabetes at age 28 years.
Differential diagnosis Differential diagnoses include permanent NDM, DEND syndrome (epilepsy, hypotonia, and developmental delay in addition to diabetes mellitus), intermediate DEND, and Wolcott-Rallison syndrome (see these terms) as well as all other syndromic forms of neonatal diabetes mellitus.
Transient neonatal diabetes mellitus (TNDB) is a type of diabetes that appears within the first few weeks of life but is transient; affected infants go into remission within a few months, with possible relapse to permanent diabetes in adolescence or adulthood. Affected individuals have slow growth before birth followed by hyperglycemia , dehydration and failure to thrive in infancy. Approximately 70% of cases are caused by the overactivity of certain genes in a region of the long (q) arm of chromosome 6 called 6q24. These cases are referred to as 6q24-related TNDB ; most (but not all) of these cases are not inherited. Other genetic causes include mutations in the KCNJ11 and ABCC8 genes, which usually cause permanent neonatal diabetes .
6q24-related transient neonatal diabetes mellitus is a type of diabetes that occurs in infants. This form of diabetes is characterized by high blood sugar levels (hyperglycemia) resulting from a shortage of the hormone insulin . Insulin controls how much glucose (a type of sugar) is passed from the blood into cells for conversion to energy. People with 6q24-related transient neonatal diabetes mellitus experience very slow growth before birth (severe intrauterine growth retardation). Affected infants have hyperglycemia and an excessive loss of fluids (dehydration), usually beginning in the first week of life.
Clinical Characteristics Clinical Description Males IPEX syndrome is generally considered to be a syndrome of neonatal enteropathy [Ruemmele et al 2004] and neonatal polyendocrinopathy [Dotta & Vendrame 2002] found in males. ... Enteropathy. The enteropathy of IPEX syndrome, often the first sign, is present in virtually all affected individuals. ... In addition, within the cohort of affected individuals with extremely early onset of symptoms (<24 h of life), the types of variants and their position within the gene vary [Reichert et al 2016]. Nomenclature IPEX syndrome may also be referred to as X-linked autoimmunity-allergic dysregulation (XLAAD) syndrome or X-linked syndrome of polyendocrinopathy, immune dysfunction, and diarrhea (XPID). Prevalence IPEX syndrome is rare: fewer than 300 affected individuals have been identified worldwide. ... Treatment of Manifestations in Individuals with IPEX Syndrome View in own window Manifestation Treatment Considerations/Other Enteropathy of IPEX syndrome Monitor fluid intake to assure adequate intravascular volume.
Some of the features were similar to those of Omenn syndrome (603554), but diabetes does not occur in that disorder which is inherited as an autosomal recessive. ... Eisenbarth and Gottlieb (2004) compared the features of 3 autoimmune polyendocrine syndromes: autoimmune polyendocrine syndrome type I (240300); autoimmune polyendocrine syndrome type II, or Schmidt syndrome (269200); and X-linked polyendocrinopathy with immune dysfunction and diarrhea. ... Unexplained rapidly progressive hemophagocytic syndrome, which proved fatal, occurred 29 months after bone marrow transplantation, when the child was nearly 3 years of age. ... Mapping Shigeoka et al. (1993) reported that the locus for this disorder maps to Xp11.2, where the Wiskott-Aldrich syndrome gene (WAS; 301000) had been assigned. ... Chatila et al. (2000) incorrectly referred to this as a 'disregulation' syndrome. They presumably meant 'dysregulation' syndrome.
Immunodysregulation polyendocrinopathy enteropathy x-linked (IPEX) syndrome is a rare autoimmune disease. it affects only males and starts in the first six months of life. The symptoms of IPEX syndrome include severe diarrhea , diabetes , skin conditions (such as eczema , erythroderma , or psoriasis ), and thyroid disease ( thyroiditis ). IPEX syndrome is caused by changes (mutations) of the FOXP3 gene, which is located on the X chromosome. There are several other diseases that are very similar to the IPEX syndrome, caused by mutations in other genes and that affect both males and females. Treatment of IPEX syndrome consists of medications that limit immune system function; a bone marrow transplantation is the only treatment that can cure the disease, but it may have several complications.
IPEX syndrome can be life-threatening in early childhood. ... Other skin disorders that cause similar symptoms are sometimes present in IPEX syndrome. The term polyendocrinopathy is used in IPEX syndrome because individuals can develop multiple disorders of the endocrine glands. ... Frequency IPEX syndrome is a rare disorder that affects an estimated 1 in 1.6 million people. ... Normal body tissues and organs are attacked, causing the multiple autoimmune disorders that develop in people with IPEX syndrome. Learn more about the gene associated with Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome FOXP3 Inheritance Pattern IPEX syndrome is inherited in an X-linked recessive pattern. ... Such conditions are classified as IPEX-like syndromes.
Epidemiology Immune dysregulation-polyendocrinopathy-enteropathy-X-linked (IPEX) syndrome prevalence is unknown. The disease has probably been underestimated, and milder clinical phenotypes surviving to adult life are being described. Clinical description IPEX syndrome most commonly develops during the first few days or weeks of life and affects exclusively boys. ... Neurological findings including peripheral neuropathy, myopathy and hypotonic are described, Etiology IPEX syndrome is caused by mutations in the FOXP3 gene (Xp11.23). ... Differential diagnosis Differential diagnosis includes Wiskott-Aldrich and Omenn syndromes, susceptibility to viral and mycobacterial infections, CD25 deficiency, IL10R deficiency, STAT5b deficiency, transient neonatal diabetes, severe combined immunodeficiency or intermediate forms of combined immunodeficiency, X-linked thrombocytopenia and pancreatic hypoplasia or agenesis.
Neufeld et al. (1980) recognized 3 types of the polyglandular autoimmune syndrome. Neufeld et al. (1981) collated information on 295 patients with autoimmune Addison disease as part of a polyglandular autoimmune syndrome. ... McKusick (1985) observed achalasia in this syndrome. The association is observed also in the achalasia-addisonianism-alcrima syndrome (231550). ... Thus, linkage studies demonstrated that the condition previously called polyglandular deficiency syndrome, Persian-Jewish type, is the same as APECED. Eisenbarth and Gottlieb (2004) compared the features of 3 autoimmune polyendocrine syndromes: autoimmune polyendocrine syndrome type I, autoimmune polyendocrine syndrome type II, and X-linked polyendocrinopathy with immune dysfunction and diarrhea (304790). ... Cetani et al. (2001) identified an Italian family with autoimmune polyendocrinopathy syndrome and a pattern of inheritance suggestive of a dominant mechanism (see MOLECULAR GENETICS).
Autoimmune polyglandular syndrome type 1 is an inherited autoimmune condition that affects many of the body's organs. Symptoms often begin in childhood or adolescence and may include mucocutaneous candidiasis , hypoparathyroidism , and Addison disease . This syndrome can cause a variety of additional signs and symptoms, such as weak teeth (enamel hypoplasia) and chronic diarrhea or constipation. ... Most people with APS-1, develop earlier and more severe symptoms than people with a related disease known as autoimmune polyendocrine syndrome type 2 (APS-2).
Specialty Medical genetics Aicardi syndrome is a rare genetic malformation syndrome characterized by the partial or complete absence of a key structure in the brain called the corpus callosum , the presence of retinal abnormalities, and seizures in the form of infantile spasms . [2] Aicardi syndrome is theorized to be caused by a defect on the X chromosome as it has thus far only been observed in girls or in boys with Klinefelter syndrome . ... The few boys that have been identified with Aicardi syndrome have proved to have 47 chromosomes including an XXY sex chromosome complement, a condition called Klinefelter syndrome . [ citation needed ] All cases of Aicardi syndrome are thought to be due to new mutations . ... "Aicardi Syndrome". Archives of Neurology . 60 (10): 1471–3. doi : 10.1001/archneur.60.10.1471 . ... PMID 19706204 . ^ a b Aicardi, Jean (January 1999). "Aicardi Syndrome: Old and New Findings" (PDF) . ... Electroenceph Clin Neurophysiol 1965; 19: 609–610 External links [ edit ] GeneReviews/NCBI/NIH/UW entry on Aicardi Syndrome OMIM entries on Aicardi syndrome Classification D ICD - 10 : Q04.0 ICD - 9-CM : 742.2 OMIM : 304050 MeSH : D058540 DiseasesDB : 29761 SNOMED CT : 80651009 External resources MedlinePlus : 001664 eMedicine : ped/58 GeneReviews : Aicardi Syndrome Orphanet : 50 v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia
Aicardi syndrome is a rare neurological disorder. The severity of the syndrome and the associated signs and symptoms vary from person to person. ... Other brain malformations such as a very small head (microcephaly) Other eye defects, such as very small eyes ( microphthalmia ) or a defect of the nerve connecting the retina to the brain ( optic nerve ) known as coloboma . The cause of Aicardi syndrome is currently unknown. Because the syndrome almost only affects females, it is believed to be caused by a change (mutation) in a gene located on the X-chromosome and inherited in a dominant X-linked manner. However, most cases are de novo , which means the genetic change happened by mistake during the making of the egg or the sperm and there are no other cases of the syndrome in the family. While there is no known cure for Aicardi syndrome, there are treatments that can help control symptoms. ... The life span of girls with Aicardi syndrome usually averages between 8 and 18 years, but several women with milder symptoms have lived into their 30’s and 40’s.
Summary Clinical characteristics. Aicardi syndrome is a neurodevelopmental disorder that affects primarily females. ... Diagnosis/testing. The diagnosis of Aicardi syndrome is based exclusively on clinical findings. ... The diagnosis of Aicardi syndrome is based exclusively on clinical findings. ... Prevalence Aicardi syndrome is very rare and appears to affect all ethnicities equally. ... In contrast to AIC, in which chorioretinal lacunae are central & optic nerves almost always involved Syndromes of unknown cause Orofaciodigital syndrome type IX (OFD9; OMIM 258865).
Aicardi syndrome is a disorder that occurs almost exclusively in females. ... Skewed X-inactivation has been identified in girls with Aicardi syndrome, further supporting the idea that the disorder is caused by a mutation in a gene on the X chromosome. However, this gene has not been identified, and it is unknown how the genetic change that causes Aicardi syndrome results in the various signs and symptoms of this disorder. ... The disorder is believed to result from new gene mutations. Aicardi syndrome is classified as an X-linked dominant condition. ... Males with a 47,XXY chromosome pattern also have a condition called Klinefelter syndrome.
Description Aicardi syndrome is characterized by a triad of callosal agenesis, infantile spasms, and chorioretinal lacunae ('holes'). ... Sutton et al. (2005) concluded that Aicardi syndrome has a distinctive facial phenotype. ... Neidich et al. (1988) found 2 new patients with Aicardi syndrome and Xp22 abnormalities. They stated that all patients have been either XX female or 47,XXY Klinefelter syndrome. ... Inheritance The inheritance of Aicardi syndrome is probably X-linked dominant with lethality in the hemizygous male. ... They found that 3 of the 7 cytogenetically normal girls with Aicardi syndrome had profoundly skewed X inactivation in their lymphocytes, supporting the concept that Aicardi syndrome is X-linked.
Epidemiology About 200 individuals with Aicardi syndrome have been reported in North America and Europe, but the true prevalence is unknown. ... Etiology It is believed that Aicardi syndrome is a sporadic disorder caused by heterozygous pathogenic variants in an X-linked gene in females, with early embryonic lethality in hemizygous males; however, a candidate region on the X chromosome has not yet been identified due to the sporadic nature of the condition. ... Differential diagnosis Differential diagnosis includes microcephaly-chorioretinopathy with or without lymphedema (in this disorder, the lacunae are peripheral and the microcephaly is significant ; girls with Aicardi syndrome typically do not have microcephaly), oculocerebrocutaneous syndrome, neuronal migration disorders and numerous syndromes that present with one or more of the features characteristic for Aicardi syndrome. Antenatal diagnosis Prenatal diagnosis may be suspected based upon fetal brain imaging. Genetic counseling Aicardi syndrome appears to be a de novo , X-linked dominant disorder with lethality in males. ... Prognosis Difficult-to-control seizures and significant neurodevelopmental disorders are the primary challenge for individuals with Aicardi syndrome. Most are not able to sit independently, walk or speak.
Popliteal pterygium syndrome is a condition that affects the development of the face, skin, and genitals. ... In some cases, people with popliteal pterygium syndrome have missing teeth. Individuals with popliteal pterygium syndrome may be born with webs of skin on the backs of the legs across the knee joint, which may impair mobility unless surgically removed. ... The average IQ of individuals with popliteal pterygium syndrome is not significantly different from that of the general population. Frequency Popliteal pterygium syndrome is a rare condition, occurring in approximately 1 in 300,000 individuals. ... Mutations in the IRF6 gene that cause popliteal pterygium syndrome may change the transcription factor's effect on the activity of certain genes.
A rare genetic, multiple congenital anomalies syndrome characterized by cleft lip, with or without cleft palate, pits in the lower lip, contractures of the lower extremities, abnormal external genitalia, syndactyly of fingers and/or toes, and a pyramidal skin fold over the hallux nail. Epidemiology The prevalence of autosomal-dominant-popliteal pterygium syndrome (AD-PPS) is unknown. However, based on the occurence of cleft lip/palate in the general population, the prevalence at birth of popliteal pterygium syndrome (PPS) has been suggested at 1/300,000 with more than 200 cases of AD-PPS being reported worldwide. ... Differential diagnosis Mildly affected AD-PPS patients have significant clinical overlap with Van der Woude syndrome (VWS), a disorder caused by deletions and mutations in the same gene ( IRF6 ). ... The cause of this variable expressivity is not known. Bartsocas-Papas syndrome, CHAND syndrome and multiple pterygium syndrome should also be considered.
These are feature of the lip-pit or Van der Woude syndrome-1 (VWS; 119300), which had been mapped to 1q32. ... Soekarman et al. (1995) described 2 families in which the popliteal pterygium syndrome occurred in 3 successive generations. While expression of the syndrome was relatively mild in the first and second generations, the patients in the third generation showed the full-blown syndrome. Differential diagnosis between mildly affected patients with the popliteal pterygium syndrome and those with Van der Woude syndrome is difficult and may even be impossible. ... Molecular Genetics Kondo et al. (2002) carried out direct sequence analysis of genes and presumptive transcripts in the critical region for popliteal pterygium syndrome and van der Woude syndrome (VWS1; 119300) identified by linkage analysis on 1q32.
Popliteal pterygium syndrome is a condition that affects the development of the face, skin, and genitals. ... Treatment may include reconstructive surgery for the oral defects, such as the cleft lip and cleft palate, and for other defects such as the popliteal pterygium or abnormal genitalia. Popliteal pterygium syndrome is part of the IRF6-related disorders which also include Van der Woude syndrome 2 .
{Short description|Medical condition}} Roberts syndrome Other names Hypomelia-hypotrichosis-facial hemangioma syndrome, SC syndrome (once thought to be an entirely separate disease), pseudothalidomide syndrome, Roberts-SC phocomelia syndrome, SC phocomelia syndrome, Appelt-Gerken-Lenz syndrome, RBS, SC pseudothalidomide syndrome, and tetraphocomelia-cleft palate syndrome. [1] [2] [3] [4] Specialty Medical genetics Roberts syndrome , or sometimes called pseudothalidomide syndrome , is an extremely rare autosomal recessive genetic disorder that is characterized by mild to severe prenatal retardation or disruption of cell division , leading to malformation of the bones in the skull, face, arms, and legs. ... Discovery of the syndrome [ edit ] The discovery of ESCO2 as the gene responsible for Roberts syndrome was made by studying samples from fifteen families affected by Roberts syndrome. ... Herrmann described another syndrome with very similar characteristics to Roberts syndrome. ... Today, Roberts Syndrome and Pseudothalidomide Syndrome (SC Syndrome) are considered to be the same disorder. [ citation needed ] The following is a list of all the alternate names that have been used for Roberts Syndrome: RBS Hypomelia-Hypotrichosis-Facial Hemangioma Syndrome SC Syndrome Pseudothalidomide Syndrome Roberts-SC Phocomelia Syndrome SC Phocomelia Syndrome Appelt-Gerken-Lenz Syndrome SC Pseudothalidomide Syndrome Tetraphocomelia-Cleft Palate Syndrome [2] [3] [4] References [ edit ] ^ a b c Kugler, Mary. ... Published 26 November 2008. ^ a b c d e f g Gordillo et al. "Roberts syndrome." ^ a b c d "Roberts syndrome."
Roberts syndrome (RBS) is characterized by pre- and postnatal growth retardation, severe symmetric limb reduction defects, craniofacial anomalies and severe intellectual deficit. ... Differential diagnosis Differential diagnoses include thalidomide embryopathy, and the Baller-Gerold, Cornelia de Lange and TAR syndromes (see these terms). Antenatal diagnosis When a previous child in the family is diagnosed with RBS and carries ESCO2 mutations, prenatal diagnosis can be performed through DNA analysis of chorionic villus samples.
Temtamy (1974) concluded that Roberts syndrome and the SC phocomelia syndrome (269000) are the same. ... Because of overlapping features in their patient, Waldenmaier et al. (1978) suggested that the SC phocomelia syndrome and the TAR syndrome (274000) are not separate from the Roberts syndrome. Tomkins et al. (1979) noted the uncertainty as to whether Roberts syndrome and the SC syndrome are separate entities. ... Gordillo et al. (2008) stated that Roberts syndrome and SC phocomelia were considered to be the same syndrome with varying phenotypic expression, and that they would henceforth designate all such cases of Roberts syndrome/SC phocomelia as 'RBS.' ... This was clearly an instance of Roberts syndrome. Bates (2003) reproduced the report of a case of Roberts syndrome by Francois Bouchard in 1672.
Roberts syndrome is a genetic disorder characterized by limb and facial abnormalities. ... Mild to severe intellectual impairment occurs in about half of all people with Roberts syndrome. Children with Roberts syndrome are born with abnormalities of all four limbs. ... In addition, people with Roberts syndrome may have heart, kidney, and genital abnormalities. Infants with a severe form of Roberts syndrome are often stillborn or die shortly after birth. ... A condition called SC phocomelia syndrome was originally thought to be distinct from Roberts syndrome; however, it is now considered to be a mild variant.
Although the SC syndrome is sometimes referred to as the pseudothalidomide syndrome, some cases of the Holt-Oram syndrome (142900) may be more deserving of that name (Lenz et al., 1974). ... The authors questioned the separateness of Roberts syndrome (268300) and this syndrome. By an analysis of phenotype, Herrmann and Opitz (1977) could not tell whether the SC phocomelia syndrome and the Roberts syndrome were 'due to different recessive genes, different alleles, or the same recessive gene.' Tomkins et al. (1979) noted the uncertainty as to whether Roberts syndrome and the SC syndrome are distinct entities. ... The usual absence of cleft palate in the SC syndrome may be a difference. Qazi et al. (1979) described chromosomal abnormalities like those of Roberts syndrome. ... Gordillo et al. (2008) stated that Roberts syndrome and SC phocomelia were considered to be the same syndrome with varying phenotypic expression, and that they would henceforth designate all such cases of Roberts syndrome/SC phocomelia as 'RBS.'
Roberts syndrome is a genetic disorder characterized by limb and facial abnormalities. ... Infants with a severe form of Roberts syndrome are often stillborn or die shortly after birth, while mildly affected individuals may live into adulthood.