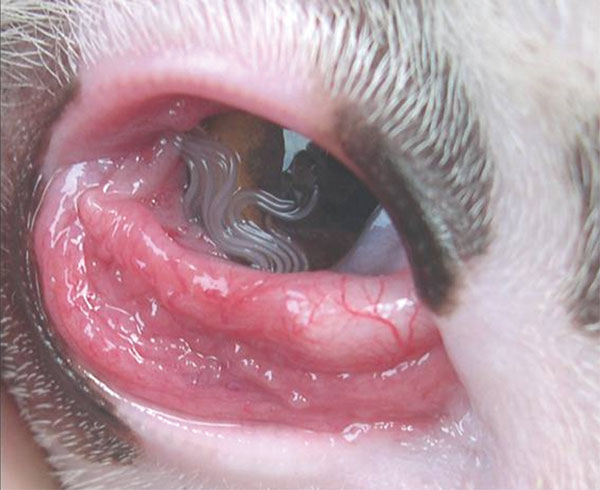
. ^ Koyama, Y; Ohira, A; Kono, T; Yoneyama, T; Shiwaku, K (2000). "Five cases of thelaziasis" . ... PMID 10936545 . ^ Dubay, SA; W, E; M, K; B, A (2000). "Bacteria and nematodes in the conjunctiva of mule deer from Wyoming and Utah" .
PMID 26506926 . ^ Tansarli, G. S.; Kostaras, E. K.; Athanasiou, S.; Falagas, M. E. (2013). ... S2CID 14514975 . ^ Donders, GG; Van Calsteren, K; Bellen, G; Reybrouck, R; Van den Bosch, T; Riphagen, I; Van Lierde, S (2009).
Emergency Medicine Clinics of North America . 23 (3): 749–70, ix. doi : 10.1016/j.emc.2005.03.001 . PMID 15982544 . ^ Crowley, R. K.; Sherlock, M.; Agha, A.; Smith, D.; Thompson, C. ... PMID 6808452 . ^ a b c d Harrington, C.; Grossman, J.; Richman, K. (2014). "Psychogenic adipsia presenting as acute kidney injury: case report and review of disorders of sodium and water metabolism in psychiatric illness".
Hypoglycemia due to endogenous insulin Congenital hyperinsulinism Transient neonatal hyperinsulinism (mechanism not known) Focal hyperinsulinism (K ATP channel disorders) Paternal SUR1 mutation with clonal loss of heterozygosity of 11p15 Paternal Kir6.2 mutation with clonal loss of heterozygosity of 11p15 Diffuse hyperinsulinism K ATP channel disorders SUR1 mutations Kir6.2 mutations Glucokinase gain-of-function mutations Hyperammonemic hyperinsulinism (glutamate dehydrogenase gain-of-function mutations) Short chain acyl coenzyme A dehydrogenase deficiency Carbohydrate-deficient glycoprotein syndrome ( Jaeken's Disease ) Beckwith-Wiedemann syndrome (suspected due to hyperinsulinism but pathophysiology uncertain: 11p15 mutation or IGF2 excess) Acquired forms of hyperinsulinism Insulinomas (insulin-secreting tumors ) Islet cell adenoma or adenomatosis Islet cell carcinoma Adult nesidioblastosis Autoimmune insulin syndrome Noninsulinoma pancreatogenous hypoglycemia Reactive hypoglycemia (also see idiopathic postprandial syndrome ) Gastric dumping syndrome Drug induced hyperinsulinism Sulfonylurea Aspirin Pentamidine Quinine Disopyramide Bordetella pertussis vaccine or infection D-chiro-inositol and myo-inositol [1] Hypoglycemia due to exogenous (injected) insulin Insulin self-injected for treatment of diabetes (i.e., diabetic hypoglycemia ) Insulin self-injected surreptitiously (e.g., Munchausen syndrome ) Insulin self-injected in a suicide attempt or fatality Various forms of diagnostic challenge or "tolerance tests" Insulin tolerance test for pituitary or adrenergic response assessment Protein challenge Leucine challenge Tolbutamide challenge Insulin potentiation therapy Insulin-induced coma for depression treatment Genetics [ edit ] There are several genetic forms of hyperinsulinemic hypoglycemia: Type OMIM Gene Locus HHF1 256450 ABCC8 11p15.1 HHF2 601820 KCNJ11 11p15.1 HHF3 602485 GCK 7p15-p13 HHF4 609975 HADH 4q22-q26 HHF5 609968 INSR 19p13.2 HHF6 606762 GLUD1 10q23.3 HHF7 610021 SLC16A1 1p13.2-p12 Diagnosis [ edit ] When the cause of hypoglycemia is not obvious, the most valuable diagnostic information is obtained from a blood sample (a "critical specimen") drawn during the hypoglycemia.
Congenital isolated hyperinsulinism (CHI), a rare endocrine disease is the most frequent cause of severe and persistent hypoglycemia in the neonatal period and early infancy and is characterized by an excessive or uncontrolled insulin secretion (inappropriate for the level of glycemia) and recurrent episodes of profound hypoglycemia requiring rapid and intensive treatment to prevent neurological sequelae. CHI comprises 2 different forms: diazoxide-sensitive diffuse hyperinsulinism and diazoxide-resistant hyperinsulinism (see these terms). Epidemiology Prevalence is estimated at 1/50,000 live births, but it may be as high as 1/2,500 in communities with substantial consanguinity. Clinical description CHI onset varies from birth through early adulthood. Neonatal onset is the most frequent; newborns, often macrosomic present with poor feeding, intolerance to fasting and persistent hypoglycemia.
Nesidioblastosis Specialty Endocrinology Nesidioblastosis is a controversial medical term for hyperinsulinemic hypoglycemia attributed to excessive insulin production by pancreatic beta cells that have an abnormal microscopic appearance. The term was coined in the first half of the 20th century. The abnormal microscopic features of the tissue included the presence of islet cell enlargement, pancreatic islet cell dysplasia , beta cells budding from ductal epithelium, and islets in close proximity to ducts. By the 1970s, nesidioblastosis was primarily used to describe the pancreatic dysfunction associated with persistent congenital hyperinsulinism and in most cases from the 1970s until the 1980s it was used as a synonym for what is now referred to as congenital hyperinsulinism. Most congenital hyperinsulinism is now known to be caused by different mechanisms than excessive proliferation of beta cells in a fetal pattern, and the term fell into disfavor after it was recognized in the late 1980s that the characteristic tissue features of nesidioblastosis were sometimes seen in pancreatic tissue from normal infants and even from adults, and are therefore not consistently associated with hyperinsulinemic hypoglycemia. In recent years, the term has been revived to describe a form of acquired hyperinsulinism with beta cell hyperplasia found in adults, especially after gastrointestinal surgery. [1] [2] [3] Evidence of mechanisms explaining the ability of weight loss surgery to induce modern-day nesidioblastosis has yet to be found; any such mechanisms are of intense interest to diabetes researchers. [ citation needed ] See also [ edit ] Congenital hyperinsulinism Neonatal hypoglycemia References [ edit ] ^ Raffel A, Krausch MM, Anlauf M, Wieben D, Braunstein S, Klöppel G, Röher H, Knoefel W (2007).
Other drugs that have been used are mycophenolate mofetil (Cellcept), azathioprine (Imuran), cyclophosphamide , rituximab , and anti-TNF therapies. [6] Hearing aids or cochlear implants may be necessary in the event of hearing loss. [6] References [ edit ] ^ Lian K, Siripurapu R, Yeung R, Hopyan J, Eng K, Aviv RI, Symons SP.
A rare systemic or rheumatologic disease characterized by the triad of central nervous system (CNS) dysfunction, branch retinal artery occlusions (BRAOs) and sensorineural hearing loss (SNHL) due to autoimmune-mediated occlusions of microvessels in the brain, retina, and inner ear. Epidemiology Susac syndrome (SuS) prevalence is still unknown. To date more than 500 cases have been reported worldwide. Young females (20-40 years) are more affected (female: male ratio 3.5:1). The age at onset ranges from 8 to 72 years (mean age: 32 years). Clinical description Characteristic is a triad of encephalopathy (cognitive and behavioral disturbances, personality changes, psychosis, preceding headaches) and/or focal CNS dysfunction, visual dysfunction due to BRAO and SNHL. The components of the triad may not be concomitantly present and may develop successively.
Susac syndrome is an autoimmune condition that affects the very small blood vessels in the brain, retina , and inner ear ( cochlea ). The condition is characterized by three main symptoms: brain disease (encephalopathy), hearing loss, and vision loss. Some people may not have all signs of Susac syndrome, but instead develop only specific combinations of the symptoms. Susac syndrome affects women more than men. The age at which symptoms begin is usually between 20 and 40 years, but some people have symptoms earlier or later than this age range. The cause of Susac syndrome is still unknown. Diagnosis is based on a clinical exam and imaging tests to look for the specific signs of Susac syndrome.
PMID 16210113 . ^ a b c d e f g h i j k l m n o p Tapsall (2001) Antimicrobial resistance in Niesseria gonorrhoeae . World Health Organization. ^ Deguchi T, Nakane K, Yasuda M, Maeda S (September 2010).
Many women who have marsupialization done find that the recurrences may slow, but do not actually stop. [ citation needed ] Epidemiology [ edit ] Two percent of women will have a Bartholin's gland cyst at some point in their lives. [2] They occur at a rate of 0.55 per 1000 person-years and in women aged 35–50 years at a rate of 1.21 per 1000 person-years. [14] The incidence of Bartholin duct cysts increases with age until menopause , and decreases thereafter. [14] Hispanic women may be more often affected than white women and black women. [2] The risk of developing a Bartholin's gland cyst increases with the number of childbirths. [2] References [ edit ] ^ a b c d e f g h i j k "Bartholin Gland Cysts" . Merck Manuals Professional Edition . Retrieved 12 September 2018 . ^ a b c d e f g h i j k l m n o p q r s t Omole, Folashade; Simmons, Barbara J.; Hacker Yolanda (2003).
Overview The Bartholin's (BAHR-toe-linz) glands are located on each side of the vaginal opening. These glands secrete fluid that helps lubricate the vagina. Sometimes the openings of these glands become obstructed, causing fluid to back up into the gland. The result is relatively painless swelling called a Bartholin's cyst. If the fluid within the cyst becomes infected, you may develop a collection of pus surrounded by inflamed tissue (abscess). A Bartholin's cyst or abscess is common. Treatment of a Bartholin's cyst depends on the size of the cyst, how painful the cyst is and whether the cyst is infected.
Koenig R.; Bach A.; Ulrike W.; Grzeschik K-H; Fuchs S. (2002). "Spectrum of the acrocallosal syndrome". ... PMID 23125460 . Putoux A.; Thomas S.; Coene K. L. M.; Davis E. E.; Alanay Y.; Ogur G.; et al. (2011).
One of the less invasive options may be preferred in a patient with many comorbidities, who is at high risk for surgery. [3] See also [ edit ] Intradural pseudoaneurysm References [ edit ] ^ Kouvelos, G. N.; Papas, N. K.; Arnaoutoglou, E. M.; Papadopoulos, G. ... P.; Landau, D. S.; Tassiopoulos, A. K. (2012). "Results of a New Human Recombinant Thrombin for the Treatment of Arterial Pseudoaneurysm".
You can help by adding to it . ( July 2017 ) History [ edit ] 1964 – GILLESPIE FD first described in two siblings with aniridia, cerebellar ataxia, and mental retardation. [1] 1971 – Sarsfield, J. K. described more cases in a family with normal NCV and muscle biopsy. [12] 1997 – Nelson J reported diffuse MRI abnormality in Cerebral and cerebellar atrophy with white matter changes suggested more diffuse disease. [6] 1998 – Dollfus H reported a patient with a phenotype suggestive of a chromosomal abnormality. [7] 2008 – Mariën P found limited cognitive deficit that closely resembles the "cerebellar cognitive and affective syndrome" (CeCAS). [9] References [ edit ] ^ a b c Gillespie, FD (Mar 1965). ... PMID 17287663 . ^ a b c d Kieslich, M; Vanselow, K; Wildhardt, G; Gebhardt, B; Weis, R; Böhles, H (Apr 2001).
A number sign (#) is used with this entry because of evidence that Gillespie syndrome (GLSP) is caused by heterozygous mutation in the ITPR1 gene (147265) on chromosome 3p26. Some patients have been reported with homozygous or compound heterozygous mutation in the ITPR1 gene. Description Gillespie syndrome is usually diagnosed in the first year of life by the presence of fixed dilated pupils in a hypotonic infant. Affected individuals have a characteristic form of iris hypoplasia in which the pupillary border of the iris exhibits a scalloped or 'festooned' edge, with iris strands extending onto the anterior lens surface at regular intervals. The key extraocular features of Gillespie syndrome are congenital hypotonia, progressive cerebellar hypoplasia, and ataxia, as well as variable cognitive impairment that is usually mild (summary by Gerber et al., 2016 and McEntagart et al., 2016).
A rare, congenital, neurological disorder characterized by the association of partial bilateral aniridia with non-progressive cerebellar ataxia, and intellectual disability. Epidemiology To date, less than 30 patients have been reported in the literature. Clinical description Aniridia is visible at birth as fixed dilated pupils and is associated with photobia. It can be accompanied with additional ocular findings such as foveal, patchy iris and/or optic nerve hypoplasia, retinal hypopigmentation, and/or pigmentary macular changes leading to reduced visual acuity. Cataract and corneal opacities are never observed. Non-progressive cerebellar ataxia is associated with delayed developmental milestones and hypotonia (visible from the first year of life), gait and balance disorders with incoordination, intention tremor, and scanning speech.
Gillespie syndrome is a disorder that involves eye abnormalities, weak muscle tone from birth (congenital hypotonia), problems with balance and coordinating movements (ataxia), and mild to moderate intellectual disability. Gillespie syndrome is characterized by underdevelopment (hypoplasia) of the colored part of the eye (the iris). In most affected individuals, part of the iris is missing (partial aniridia) in both eyes. In addition, the irises have a characteristic uneven pattern known as "scalloping" at the inner (pupillary) edge. The pupils are enlarged (dilated) and are fixed, which means they do not get smaller (constrict) in response to light.
A number sign (#) is used with this entry because of evidence that the exclusively skeletal form of Antley-Bixler syndrome can be caused by heterozygous mutation in a fibroblast growth factor receptor gene, FGFR2 (176943), on chromosome 10q26. A form of Antley-Bixler syndrome that includes disordered steroidogenesis (ABS1; 201750) is caused by mutation in the gene encoding cytochrome P450 oxidoreductase (POR; 124015). Description The Antley-Bixler syndrome (ABS) is an exceptionally rare craniosynostosis syndrome characterized by radiohumeral synostosis present from the perinatal period. There is a wide spectrum of anomalies seen in ABS, including midface hypoplasia, choanal stenosis or atresia, and multiple joint contractures. Mortality has been reported to be as high as 80% in the neonatal period, primarily due to airway compromise, and prognosis improves with increasing age (summary by McGlaughlin et al., 2010).
Antley Bixler syndrome is a rare condition that is primarily characterized by craniofacial abnormalities and other skeletal problems. The signs and symptoms vary significantly from person to person but may include craniosynostosis; midface hypoplasia (underdeveloped middle region of the face); frontal bossing ; protruding eyes; low-set, unusually-formed ears; choanal atresia or stenosis (narrowing); fusion of adjacent arm bones (synostosis); joint contractures ; arachnodactyly ; bowing of the thigh bones; and/or urogenital (urinary tract and genital) abnormalities. The exact underlying cause of Antley Bixler syndrome is unknown in many cases; however, some are due to changes (mutations) in the FGFR2 gene or the POR gene. There appear to be autosomal dominant and autosomal recessive forms of the condition. Treatment is based on the signs and symptoms present in each person.
A very rare disorder characterised by craniosynostosis with midface hypoplasia, radiohumeral synostosis, femoral bowing and joint contractures. Epidemiology It has been described in more than 30 patients. Clinical description Children present with characteristic facial features, including a large domed forehead, flat nose, and midface hypoplasia with proptosis and dysplastic ears. Arachnodactyly and/or camptodactyly have also been reported. A diverse range of malformations (cardiac, anal or vertebral) are often associated. Urogenital anomalies with sexual ambiguity due to impaired steroidogenesis can occur. Intellectual development is variable. Differential diagnosis A similar clinical picture is observed in patients exposed in utero to fluconazole, a lanosterol 14 alpha-demethylase inhibitor.
The body lotion, prior to the event, had been used as a cheap substitute for vodka by the impoverished people in the region despite warnings on the lotion's bottles that it was not safe for drinking and long-standing problems with alcohol poisoning across the country. [16] During the COVID-19 pandemic , Iranian media reported that nearly 300 people had died and over a thousand became ill due to methanol poisoning in the belief that drinking the alcohol could help with the disease. [17] In the United States, the Food and Drug Administration discovered that a number of brands of hand sanitizer manufactured in Mexico during the pandemic contained methanol, and urged the public to avoid using the affected products. [18] See also [ edit ] Ethylene glycol poisoning References [ edit ] ^ a b c d e f g h i j k l m Kruse, JA (October 2012). "Methanol and ethylene glycol intoxication". ... PMID 22998995 . ^ a b c d e f g h i j k l m n o p q r s Beauchamp, GA; Valento, M (September 2016).
.; Treanor, John J.; Wolff, Mark; Newman, Frances K.; Atmar, Robert L.; Edelman, Robert; Nolan, Carrie M.; Belshe, Robert B.; National Institute of Allergy and Infectious Diseases Smallpox Vaccine Study Group (25 April 2002). ... PMID 11923490 . ^ Frey, Sharon E.; Newman, Frances K.; Cruz, John; Shelton, W. Brian; Tennant, Janice M.; Polach, Tamara; Rothman, Alan L.; Kennedy, Jeffrey S.; Wolff, Mark; Belshe, Robert B.; Ennis, Francis A. (25 April 2002).
Journal of Psychophysiology , 24 (4), 213-214. doi : 10.1027/0269-8803/a000034 ^ Schindler, A. G., Tsutsui, K. T. and Clark, J. J. (2014), Chronic Alcohol Intake During Adolescence, but not Adulthood, Promotes Persistent Deficits in Risk-Based Decision Making. ... Johns Hopkins . Retrieved April 20, 2016 . ^ K. S. Kendler, C. Gardner and D. M.
.; Mammel, Kathleen A.; Katzman, Debra K.; Rome, Ellen S.; Callahan, S. Todd; Malizio, Joan; Kearney, Sarah (2014-07-01). ... Scientific Proceedings of the Annual Meeting of American Academy of Child and Adolescent Psychiatry . 10 : 50. ^ Schreck KA, Williams K, Smith AF. A comparison of eating behaviors between children with and without Autism" Journal of Autism and Developmental Disabilities 2004; 34: 433-438. ^ Evans, E. (2013).
International Journal of Oral and Maxillofacial Surgery . 40 (8): 810–4. doi : 10.1016/j.ijom.2011.02.031 . PMID 21474286 . ^ Sharma, N. K., Singh, A. K., Pandey, A., Verma, V., & Singh, S. (2015).
PMID 22346281 . ^ Sibbald, R. G.; Black, A. K.; Eady, R. A.; James, M; Greaves, M. ... PMID 19130582 . ^ Zuberbier, T; Asero, R; Bindslev-Jensen, C; Walter Canonica, G; Church, M. K.; Giménez-Arnau, A; Grattan, C. E.; Kapp, A; Merk, H.
Shelley and Rawnsley (1964) described urticaria on contact with water. Heat or cold had no effect. Bonnetblanc et al. (1979) reported aquagenic urticaria in a woman and her paternal aunt. Inheritance - Autosomal dominant Skin - Urticaria on contact with water - No effect of heat or cold ▲ Close
Aquagenic urticaria is a rare condition in which urticaria (hives) develop rapidly after the skin comes in contact with water, regardless of its temperature. It most commonly affects women and symptoms often start around the onset of puberty. Some patients report itching too. It is a form of physical urticaria . The exact underlying cause of aquagenic urticaria is currently unknown. Due to the rarity of the condition, there is very limited data regarding the effectiveness of individual treatments; however, various medications and therapies have been used with variable success.
Lewis: Menstrual Health in Women's Lives. p.152 (University of Illinois Press, 1991) ISBN 0252062094 ^ The Headaches; Editors: Jes Olesen, Peer Tfelt-Hansen, et al. p.331 (Lippincott Williams & Wilkins; Third edition, 2005) ISBN 0781754003 ^ Pinkerman, B.; Holroyd, K. (2010). "Menstrual and nonmenstrual migraines differ in women with menstrually-related migraine". ... Dodick, Peter Sándor, editors: Comorbidity in Migraine; Wiley-Blackwell; 1 edition, 2011 p.vi ISBN 1405185554 ^ Tan, K. S. (2001). "Premenstrual asthma: Epidemiology, pathogenesis and treatment".