Clinical Variability Delahunt et al. (1993) reported a family in which a 27-month-old girl and a 31-month-old boy underwent nephrectomy for cystic nephroma and were free of disease in the contralateral kidney 16 and 14 years later, respectively. Their 28-month-old sister underwent pleuropneumonectomy with postoperative chemotherapy for PPB and died of recurrent disease 9 months later.
Pleuropulmonary blastoma Other names Pulmonary blastoma Specialty Oncology Pleuropulmonary blastoma ( PPB ) is a rare cancer originating in the lung or pleural cavity . It occurs most often in infants and young children [1] but also has been reported in adults. [2] In a retrospective review of 204 children with lung tumors, pleuropulmonary blastoma and carcinoid tumor were the most common primary tumors (83% of the 204 children had secondary tumors spread from cancers elsewhere in the body). [1] Pleuropulmonary blastoma is regarded as malignant . The male:female ratio is approximately one. Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 3.1 Types 4 Treatment 5 History 6 See also 7 References 8 External links Signs and symptoms [ edit ] Symptoms may include coughing , an upper respiratory tract infection , shortness of breath , and chest pain . These symptoms are very non-specific, and can be caused by other types of tumor in the lung or mediastinum more generally, and by other conditions. Imaging (X-ray, CT, MRI) may be used to determine the presence and precise location of a tumor, but not a specific diagnosis of PPB or other tumor. [3] Doctors are unable to tell if a child has PPB right away, and not upper respiratory tract infection , until more test are taken and they show that there is no infection.
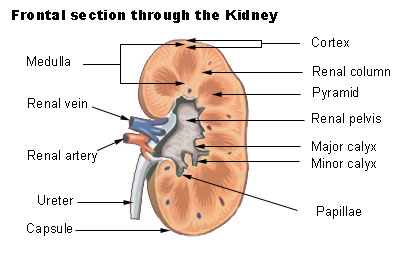
DICER1-related pleuropulmonary blastoma cancer predisposition syndrome causes a moderately increased risk for certain cancers and tumors. The lungs, kidneys, ovaries, and thyroid are the most commonly involved sites. Pleuropulmonary blastoma is the most commonly associated tumor and often occurs in infants and young children. Cysts in the kidneys (cystic nephroma) are also associated with DICER1 syndrome. These cysts typically develop in childhood, but do not usually cause any health problems.
A rare respiratory tumor characterized by an aggressive, malignant, dysontogenetic neoplasm of intrathoracic (pulmonary, pleural, or combined) mesenchyme occurring in young children. Three subtypes can be distinguished, type 1 being purely cystic, type 2 cystic and solid, and type 3 purely solid. Type 1 lesions may progress to the more malignant types 2 and 3, which are associated with central nervous system and bone metastasis. The tumor is often part of pleuropulmonary blastoma family tumor and dysplasia syndrome. It can also be associated with multilocular cystic nephroma or other neoplasms.
A number sign (#) is used with this entry because of evidence that multiple synostoses syndrome-3 (SYNS3) is caused by heterozygous mutation in the FGF9 gene (600921) on chromosome 13q12. For a general phenotypic description and a discussion of genetic heterogeneity of multiple synostoses syndrome, see SYNS1 (186500). Clinical Features Wu et al. (2009) described 12 affected individuals from a 5-generation Chinese family segregating autosomal dominant multiple synostoses syndrome, with fusions of proximal interphalangeal, carpal-tarsal, and humeroradial joints. Hearing, stature, and intelligence were normal in all affected individuals. Only mild semidislocation or cubital valgus at elbow joints or limitation of finger joint flexion was found in 4 patients aged 11 years or below, suggesting that the phenotype is age dependent.
A number sign (#) is used with this entry because of evidence that multiple synostoses syndrome-2 (SYNS2) is caused by heterozygous mutation in the GDF5 gene (601146) on chromosome 20q11. For a general phenotypic description and a discussion of genetic heterogeneity of multiple synostoses syndrome, see SYNS1 (186500). Clinical Features Akarsu et al. (1999) described a large Iranian family with tarsal-carpal coalition, humeroradial synostosis, brachydactyly, and proximal symphalangism inherited in an autosomal dominant pattern. These findings were considered consistent with the syndrome described by Pearlman (see 186400) but showed considerable overlap with other multiple synostosis syndromes. They referred to the phenotype as multiple synostosis type 2 (SYNS2).
All patients were microcephalic and had growth failure leading to proportionate short stature. At the time of the analysis, 28 individuals had died, with a mean age of death of 8.4 years with a range of 17 months to 30 years.
All patients were microcephalic and had growth failure leading to proportionate short stature. At the time of the analysis, 28 individuals had died, with a mean age of death of 8.4 years with a range of 17 months to 30 years.
Cockayne syndrome is a rare disease which causes short stature, premature aging ( progeria ), severe photosensitivity , and moderate to severe learning delay. This syndrome also includes failure to thrive in the newborn, very small head (microcephaly), and impaired nervous system development. Other symptoms may include hearing loss, tooth decay, vision problems, and bone abnormalities. There are three subtypes according to the severity of the disease and the onset of the symptoms: Cockayne syndrome type 1 (type A) , sometimes called “classic” or "moderate" Cockayne syndrome, diagnosed during early childhood Cockayne syndrome type 2 (type B) , sometimes referred to as the “severe” or "early-onset" type, presenting with growth and developmental abnormalities at birth Cockayne syndrome type 3 (type C) , a milder form of the disorder Cockayne syndrome is caused by mutations in either the ERCC8 (CSA) or ERCC6 (CSB) genes. Inheritance is autosomal recessive. Type 2 is the most severe and affected people usually do not survive past childhood.
Catastrophic antiphospholipid syndrome (CAPS) is a rare form of antiphospholipid syndrome (APS). In CAPS multiple blood clots form throughout the body over a short period of time (usually within a week). CAPS is a medical emergency, as clots can cause life-threatening multi-organ failure. The cause of CAPS is unknown. A widely accepted explanation is that it is caused by a combination of gene mutations (making one more susceptible to CAPS) and an environmental trigger, such as an infection, trauma, or surgery.
A rare systemic autoimmune disease characterized by acute onset of life-threatening thromboses in three or more organs either simultaneously or within less than a week, in the presence of serum antiphospholipid antibodies (such as lupus anticoagulant, anticardiolipin antibodies, and anti-beta2-glycoprotein 1 antibodies), and with histopathological confirmation of small-vessel occlusion in at least one affected organ. The condition occurs in a small subset of patients with antiphospholipid syndrome, often precipitated by infection, trauma, or surgery.
In all, 58% of patients with a DNMT3A mutation progressed to AML, compared to 28% without a mutation. Analysis of the bone marrow cells showed that the mutations were present in nearly all of the cells, although the myeloblast count was less than 30% for most samples, suggesting that DNMT3A mutations are very early genetic events in MDS and may confer a clonal advantage to cells with the mutation.
Overview Myelodysplastic syndromes are a group of disorders caused by blood cells that are poorly formed or don't work properly. Myelodysplastic syndromes result from something amiss in the spongy material inside your bones where blood cells are made (bone marrow). Management of myelodysplastic syndromes is most often intended to slow the disease, ease symptoms and prevent complications. Common measures include blood transfusions and medications to boost blood cell production. In certain situations, a bone marrow transplant, also known as a stem cell transplant, may be recommended to replace your bone marrow with healthy bone marrow from a donor.
Myelodysplastic syndromes (MDS) are a group of blood disorders characterized by abnormal development of blood cells within the bone marrow. People with MDS have abnormally low blood cell levels (low blood counts ). Signs and symptoms may include dizziness, fatigue, weakness, shortness of breath, bruising and bleeding, frequent infections, and headaches. In some people with MDS, the condition progresses to bone marrow failure or develops into acute leukemia . MDS develops when a cell with a mutation replicates, and the resulting copies begin to predominate in the bone marrow and suppress healthy stem cells.
Food and Drug Administration (FDA) for the treatment of MDS: 5-azacytidine : 21-month median survival [27] [28] [29] [30] Decitabine : Complete response rate reported as high as 43%. ... Journal of the American Medical Association . 152 (11): 1018–28. doi : 10.1001/jama.1953.03690110032010 .
Strangulation and necrosis commonly occur with organoaxial gastric volvulus and have been reported in 5–28% of cases. The key imaging feature of organoaxial volvulus is that the greater curvature is located above the lesser curvature of the stomach. [1] Mesenteroaxial type [ edit ] The mesenteroaxial axis bisects the lesser and greater curvatures.
A number sign (#) is used with this entry because this form of hereditary amyloidosis is caused by heterozygous mutation in the TTR gene (176300) on chromosome 18q12. Description Hereditary amyloidoses are a clinically and genetically heterogeneous group of autosomal dominantly inherited diseases characterized by the deposit of unsoluble protein fibrils in the extracellular matrix (summary by Hund et al., 2001). Patients with transthyretin amyloidosis typically present with polyneuropathy, carpal tunnel syndrome, autonomic insufficiency, cardiomyopathy, and gastrointestinal features, occasionally accompanied by vitreous opacities and renal insufficiency. In later stages of the disease severe diarrhea with malabsorption, cachexia, incapacitating neuropathy, severe cardiac disturbances, and marked orthostatic hypotension dominate the clinical picture. Death usually occurs 5 to 15 years after onset of symptoms. Before the emergence of molecular genetics, hereditary amyloidoses were classified into 4 subtypes according to symptom constellation and ethnic origin (summary by Hund et al., 2001).
Familial amyloid polyneuropathy (FAP) or transthyretin (TTR) amyloid polyneuropathy is a progressive sensorimotor and autonomic neuropathy of adulthood onset. Weight loss and cardiac involvement are frequent; ocular or renal complications may also occur. Epidemiology The prevalence worldwide is unknown, but the prevalence in the general population in Japan has recently been estimated at around 1 per million. Clinical description FAP is clinically heterogeneous, with the clinical presentation depending on the genotype and geographic origin. FAP usually presents as a length-dependent sensory polyneuropathy with autonomic disturbances.
This article may be too technical for most readers to understand . Please help improve it to make it understandable to non-experts , without removing the technical details. ( June 2009 ) ( Learn how and when to remove this template message ) Familial amyloid neuropathy Specialty Endocrinology The familial amyloid neuropathies (or familial amyloidotic neuropathies , neuropathic heredofamilial amyloidosis , familial amyloid polyneuropathy ) are a rare group of autosomal dominant diseases wherein the autonomic nervous system and/or other nerves are compromised by protein aggregation and/or amyloid fibril formation. [1] [2] [3] Contents 1 Classification 2 Treatment 3 References 4 External links Classification [ edit ] The aggregation of one precursor protein leads to peripheral neuropathy and/or autonomic nervous system dysfunction. These proteins include: transthyretin (ATTR, the most commonly implicated protein), apolipoprotein A1 , and gelsolin . [4] Due to the rareness of the other types of familial neuropathies, transthyretin amyloidogenesis-associated polyneuropathy should probably be considered first. [5] "FAP-I" and "FAP-II" are associated with transthyretin . [1] [6] ( Senile systemic amyloidosis [abbreviated "SSA"] is also associated with transthyretin aggregation.) "FAP-III" is also known as "Iowa-type", and involves apolipoprotein A1 . [7] "FAP-IV" is also known as " Finnish-type ", and involves gelsolin . [8] Fibrinogen , apolipoprotein A1 , and lysozyme are associated with a closely related condition, familial visceral amyloidosis . Diagnosis is confirmed by blood tests, organ biopsies, and tissue biopsies. Genetic testing can also be used to confirm a mutation in the TTR gene.
.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K. (2005). "Ch. 28 The central nervous system". Robbins and Cotran pathologic basis of disease (7th ed.).
Ependymoma is the most frequent intramedullary tumor in adults (but accounts for only 10-12% of pediatric central nervous system tumors), and can be benign or anaplastic. Ependymoma arise from the ependymal cells of the cerebral ventricles, corticle rests and central canal of the spinal cord, and manifest with variable symptoms such headache, vomiting, seizures, focal neurological signs and loss of vision and can cause obstructive hydrocephalus in some cases.
A tumor of neurectodermal origin arising from ependymal cells that line the ventricles and central canal of the spinal cord, that can occur in both children and adults, and that is characterized by wide a range of clinical manifestations depending on the location of the tumor, such as intracranial hypertension for tumors originating in the posterior fossa, behavioural changes and pyramidal signs for supratentorial tumors, and dysesthesia for tumors of the spinal cord. They can be classified as myxopapillary ependymoma, subependymoma, ependymoma (low grade tumors) or anaplastic ependymoma (grade III tumors).
"Sensory Reinforcement of Eyeblink Rate in a Decorticate Human", American Journal of Mental Deficiency , 80, 6, 665-7, May 1976 ^ Rare Disease Day: February 28. "What is a Rare Disease?" . Rare Disease .
A rare cerebral malformation characterized by an almost or complete lack of cortex, specifically the cerebral hemispheres, with the cranium and meninges completely intact. In most cases, death occurs in utero or in the first weeks of life. Developmental delay, drug-resistant seizures, spastic diplegia, severe growth failure, deafness and blindness are typical.
Hydranencephaly is a rare condition in which the brain's cerebral hemispheres are absent and replaced by sacs filled with cerebrospinal fluid (CSF) . Affected infants may appear and act normal at birth, but irritability and hypertonia often develop within a few weeks. Other signs and symptoms may include seizures, hydrocephalus, visual impairment, lack of growth, deafness, blindness, paralysis, and intellectual disabilities. Prognosis is typically poor with many affected children dying before one year of age. In rare cases, children may survive for several years or more. It has been suspected to be an inherited condition, although some researchers believe it may be caused by prenatal blockage of the carotid artery where it enters the cranium.
A number sign (#) is used with this entry because Rotor type hyperbilirubinemia (HBLRR) is caused by digenic inheritance of homozygous mutations in the SLCO1B1 (604843) and SLCO1B3 (605495) genes, which are located near each other on chromosome 12p. Description The Rotor type of hyperbilirubinemia is an autosomal recessive form of primary conjugated hyperbilirubinemia. It is similar to Dubin-Johnson syndrome (DJS; 237500) in that affected individuals develop mild jaundice not associated with hemolysis shortly after birth or in childhood. However, Rotor syndrome can be distinguished from DJS by a lack of hepatocyte pigment deposits, delayed plasma clearance of the unconjugated anionic dye bromsulfthalein, poor hepatic visualization on certain radiographic imaging studies, and prominent urinary excretion of coproporphyrin I (summary by van de Steeg et al., 2012). Clinical Features Because of clinical similarities, the Rotor and Dubin-Johnson syndromes were initially considered to be the same entity.
A benign, inherited liver disorder characterized by chronic, predominantly conjugated, nonhemolytic hyperbilirubinemia with normal liver histology. Epidemiology Rotor syndrome (RT) is a very rare disorder: the exact prevalence is unknown but over 50 cases have been reported in literature so far. Clinical description It is usually diagnosed in children or adolescents, but mild jaundice is often noted from birth. The main symptom consists of mild-to-moderate, recurrent jaundice without pruritus. Attacks of abdominal pain and low-grade fever can occur but are rare.
Summary Clinical characteristics. Rotor syndrome is characterized by mild conjugated and unconjugated hyperbilirubinemia that usually begins shortly after birth or in childhood. Jaundice may be intermittent. Conjunctival icterus may be the only clinical manifestation. Diagnosis/testing. The diagnosis of Rotor syndrome is established in a proband with isolated, predominantly conjugated hyperbilirubinemia without cholestasis or liver injury and typical findings on cholescintigraphy. Identification of biallelic pathogenic variants in SLCO1B1 and SLCO1B3 on molecular genetic testing can confirm the diagnosis when cholescintigraphy is either not available or not recommended due to risks associated with the procedure. Management. Treatment of manifestations: No treatment required. Agents/circumstances to avoid: Although no adverse drug effects have been documented in persons with Rotor syndrome, the absence of the hepatic proteins OATP1B1 and OATP1B3 may have serious consequences for liver uptake – and thus for the toxicity of numerous commonly used drugs and/or their metabolites.
Rotor syndrome is an inherited disorder characterized by elevated levels of bilirubin in the blood (hyperbilirubinemia). Bilirubin is produced when red blood cells are broken down, and has an orange-yellow tint. The buildup of bilirubin in the body causes yellowing of the skin or whites of the eyes (jaundice), which is the only symptom of the disorder. Jaundice is usually evident in infancy or early childhood, and it may come and go. Rotor syndrome is caused by having mutations in both the SLCO1B1 and SLCO1B3 genes and is inherited in an autosomal recessive manner.
Rotor syndrome is a relatively mild condition characterized by elevated levels of a substance called bilirubin in the blood (hyperbilirubinemia). Bilirubin is produced when red blood cells are broken down. It has an orange-yellow tint, and buildup of this substance can cause yellowing of the skin or whites of the eyes (jaundice). In people with Rotor syndrome, jaundice is usually evident shortly after birth or in childhood and may come and go; yellowing of the whites of the eyes (also called conjunctival icterus) is often the only symptom. There are two forms of bilirubin in the body: a toxic form called unconjugated bilirubin and a nontoxic form called conjugated bilirubin. People with Rotor syndrome have a buildup of both unconjugated and conjugated bilirubin in their blood, but the majority is conjugated.
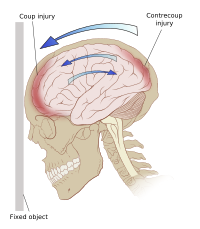
. ^ Transcript ^ Transcript ^ Archive of "The Next Big Thing" radio, Jan. 13, 2005 , Perry Mason clip played at 23:43 and 28:25. External links [ edit ] The dictionary definition of contrecoup at Wiktionary
"Homophily or the Queen Bee Syndrome: Female Evaluation of Female Leadership" . Small Group Research . 28 (4): 483–499. doi : 10.1177/1046496497284001 .
Health Essentials from Cleveland Clinic . 2014-07-28 . Retrieved 2020-11-02 . ^ "Delayed Speech or Language Development (for Parents) - Nemours KidsHealth" . kidshealth.org .