-
Pigmentation Disorder
Wikipedia
External links [ edit ] Classification D ICD - 10 : L80 - L81 ICD - 9-CM : 709.01 MeSH : D010859 v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark This cutaneous condition article is a stub .
-
Bonnet–dechaume–blanc Syndrome
Wikipedia
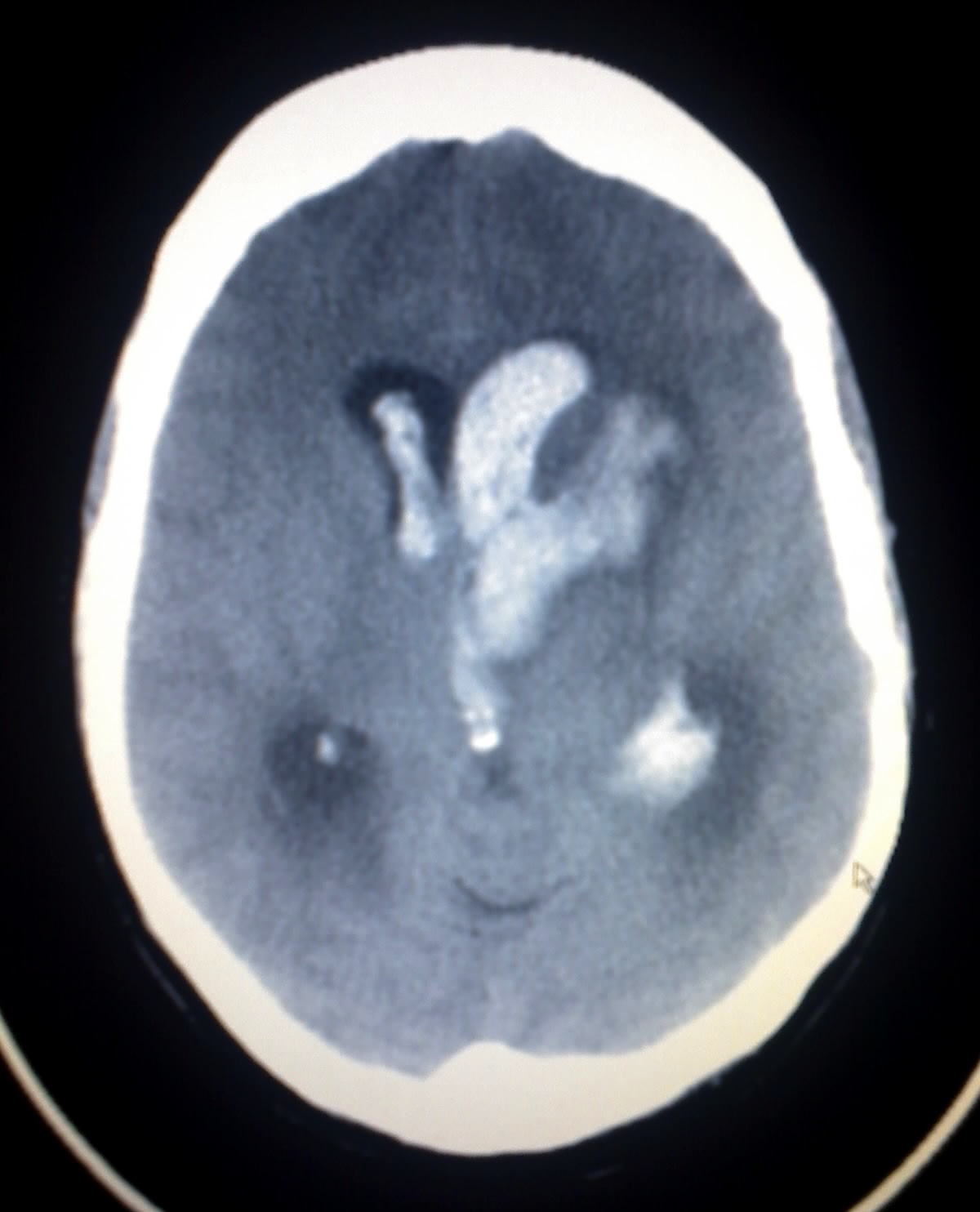
Bonnet–Dechaume–Blanc syndrome Other names Arteriovenous aneurysm of mid-brain and retina, facial nevi and mental changes, Cerebrofacial arteriovenous metameric syndrome type 2 CT scan showing intracranial hemorrhage Specialty Medical genetics Bonnet–Dechaume–Blanc syndrome , also known as Wyburn-Mason syndrome , is a rare congenital disorder characterized by arteriovenous malformations of the brain , retina or facial nevi . [1] The syndrome has a number of possible symptoms and can, more rarely, affect the skin , bones , kidneys , muscles , and gastrointestinal tract . [2] When the syndrome affects the brain, people can experience severe headaches, seizures, acute stroke, meningism , and progressive neurological deficits due to acute or chronic ischaemia caused by arteriovenous shunting. [2] [3] In the retina, the syndrome causes retinocephalic vascular malformations that tend to be present with intracranial hemorrhage and lead to decreased visual acuity, proptosis , pupillary defects, optic atrophy, congestion of bulbar conjunctiva , and visual field defects. [4] [5] Retinal lesions can be unilateral and tortuous, and symptoms begin to appear in the second and third decades of life. [4] The syndrome can present with cutaneous lesions, or skin with different texture, thickness, and color, usually on the face. [5] The facial features caused by the syndrome vary from slight discoloration to extensive nevi and angiomas of the skin. [3] In some cases, the frontal and maxillary sinus may also be affected. [5] There have only been 52 reported cases of patients with Bonnet–Dechaume–Blanc syndrome as of 2012. [2] Symptoms are rarely noticed in children and the syndrome is often diagnosed in late childhood or early adulthood when visual impairment is noticed. [3] Fluorescein angiography is commonly used to diagnose the syndrome. [6] There have been several methods in treating patients with Bonnet–Dechaume–Blanc syndrome. ... Bonnet–Dechaume–Blanc syndrome results from arteriovenous malformations . ... A. (18 January 2007). "A case report of Wyburn-Mason syndrome and review of the literature". ... "The congenital unilateral retinocephalic vascular malformation syndrome (bonnet-dechaume-blanc syndrome or wyburn-mason syndrome): review of the literature". ... External links [ edit ] Classification D ICD - 10 : Q28.2 MeSH : C536752 External resources Orphanet : 53719 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome
-
Snyder–robinson Syndrome
Wikipedia
The syndrome has also been referred to as Snyder–Robinson X-linked mental retardation syndrome ( MRXSSR ) and spermine synthase deficiency . ... Snyder [10] and Arthur Robinson, who described the syndrome as "recessive sex-linked mental retardation in the absence of other recognizable abnormalities". [11] References [ edit ] ^ a b c "Snyder-Robinson syndrome" . ... (June 27, 2013). "Snyder-Robinson Syndrome" . GeneReviews . Seattle: University of Washington, Seattle . ... (June 27, 2013). "Snyder-Robinson Syndrome" . GeneReviews . Seattle: University of Washington, Seattle . ... Classification D ICD - 10 : Q87.8 OMIM : 309583 MeSH : C536678 DiseasesDB : 35186 SNOMED CT : 702416008 External resources GeneReviews : NBK144284 Orphanet : 3063 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome
-
Townes–brocks Syndrome
Wikipedia
Townes–Brocks syndrome This condition is inherited in an autosomal dominant manner Specialty Medical genetics Townes–Brocks syndrome [1] ( TBS ) is a rare genetic disease that has been described in approximately 200 cases in the published literature. ... The clinical features of TBS overlap with VATER and VACTERL associations, oculo-auriculo-vertebral (OAV) spectrum , branchio-oto-renal (BOR) syndrome, and Fanconi anemia and other 'anus-hand-ear' syndromes. [4] Although some symptoms can be life-threatening, many people diagnosed with Townes-Brocks Syndrome live a normal lifespan. [2] Diagnosis [ edit ] This section is empty. ... ISBN 978-1-4160-2999-1 . ^ a b c Contact a Family Archived 2006-09-24 at the Wayback Machine ^ National Organization for Rare Diseases ^ GeneDX Archived 2006-10-16 at Archive.today External links [ edit ] GeneReview/NCBI/NIH/UW entry on Townes-Brocks Syndrome Classification D ICD - 10 : Q87.8 OMIM : 107480 MeSH : C536974 DiseasesDB : 32163 SNOMED CT : 24750000 External resources Orphanet : 857 v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions )

-
Brainstem Stroke Syndrome
Wikipedia
Brainstem stroke Specialty Neurology A brainstem stroke syndrome falls under the broader category of stroke syndromes , or specific symptoms caused by vascular injury to an area of brain (for example, the lacunar syndromes ). ... A more serious outcome is locked-in syndrome . Contents 1 Classic Syndromes 2 History 2.1 Kate Allatt 2.2 Jean-Dominique Bauby 2.3 Rabbi Ronnie Cahana 2.4 Tony Nicklinson 2.5 Julia Tavalaro 3 See also 4 References 5 External links Classic Syndromes [ edit ] The midbrain syndromes (Significant overlap between these three syndromes) Superior alternating hemiplegia or Weber's syndrome Paramedian midbrain syndrome or Benedikt's syndrome Claude's syndrome Medial pontine syndrome or Middle alternating hemiplegia or Foville's syndrome Lateral pontine syndrome or Marie-Foix syndrome Medial medullary syndrome or Inferior alternating hemiplegia Lateral medullary syndrome or Wallenberg syndrome History [ edit ] A history of locked in syndromes. ... She has successfully recovered from locked-in syndrome. Now she runs Fighting Strokes , and devotes her life to assisting those with locked-in syndrome. [ citation needed ] Jean-Dominique Bauby [ edit ] Parisian journalist Jean-Dominique Bauby suffered a stroke in December 1995, and, when he awoke 20 days later, he found his body was almost completely paralyzed; he could control only his left eyelid. ... She died in 2003 at the age of 68. [7] [8] See also [ edit ] Alternating hemiplegia Lacunar syndromes Posterior cerebral artery syndrome Middle cerebral artery syndrome Anterior cerebral artery syndrome References [ edit ] ^ "The Diving Bell And The Butterfly" . ... External links [ edit ] Classification D ICD - 10 : G46.3 ICD - 9-CM : 434.91 v t e Cerebrovascular diseases including stroke Ischaemic stroke Brain Anterior cerebral artery syndrome Middle cerebral artery syndrome Posterior cerebral artery syndrome Amaurosis fugax Moyamoya disease Dejerine–Roussy syndrome Watershed stroke Lacunar stroke Brain stem Brainstem stroke syndrome Medulla Medial medullary syndrome Lateral medullary syndrome Pons Medial pontine syndrome / Foville's Lateral pontine syndrome / Millard-Gubler Midbrain Weber's syndrome Benedikt syndrome Claude's syndrome Cerebellum Cerebellar stroke syndrome Extracranial arteries Carotid artery stenosis precerebral Anterior spinal artery syndrome Vertebrobasilar insufficiency Subclavian steal syndrome Classification Brain ischemia Cerebral infarction Classification Transient ischemic attack Total anterior circulation infarct Partial anterior circulation infarct Other CADASIL Binswanger's disease Transient global amnesia Haemorrhagic stroke Extra-axial Epidural Subdural Subarachnoid Cerebral/Intra-axial Intraventricular Brainstem Duret haemorrhages General Intracranial hemorrhage Aneurysm Intracranial aneurysm Charcot–Bouchard aneurysm Other Cerebral vasculitis Cerebral venous sinus thrombosis v t e Symptoms , signs and syndromes associated with lesions of the brain and brainstem Brainstem Medulla (CN 8, 9, 10, 12) Lateral medullary syndrome/Wallenberg PICA Medial medullary syndrome/Dejerine ASA Pons (CN 5, 6, 7, 8) Upper dorsal pontine syndrome/Raymond-Céstan syndrome Lateral pontine syndrome ( AICA ) (lateral) Medial pontine syndrome / Millard–Gubler syndrome / Foville's syndrome ( basilar ) Locked-in syndrome Internuclear ophthalmoplegia One and a half syndrome Midbrain (CN 3, 4) Weber's syndrome ventral peduncle, PCA Benedikt syndrome ventral tegmentum, PCA Parinaud's syndrome dorsal, tumor Claude's syndrome Other Alternating hemiplegia Cerebellum Latearl Dysmetria Dysdiadochokinesia Intention tremor ) Medial Cerebellar ataxia Basal ganglia Chorea Dystonia Parkinson's disease Cortex ACA syndrome MCA syndrome PCA syndrome Frontal lobe Expressive aphasia Abulia Parietal lobe Receptive aphasia Hemispatial neglect Gerstmann syndrome Astereognosis Occipital lobe Bálint's syndrome Cortical blindness Pure alexia Temporal lobe Cortical deafness Prosopagnosia Thalamus Thalamic syndrome Other Upper motor neuron lesion Aphasia
-
Jacobsen Syndrome
Wikipedia
Jacobsen syndrome Other names Del(11)(qter), distal deletion 11q, distal monosomy 11q, monosomy 11qter Girl with Jacobsen syndrome Specialty Medical genetics Jacobsen syndrome is a rare chromosomal disorder resulting from deletion of genes from chromosome 11 that includes band 11q24.1. ... American Journal of Medical Genetics Part C: Seminars in Medical Genetics . 169 (3): 239–250. doi : 10.1002/ajmg.c.31448 . PMID 26285164 . ^ "Jacobsen Syndrome" . DoveMed . ^ "11q deletion syndrome" . www.socialstyrelsen.se (in Swedish). ^ "Jacobsen Syndrome" . ... American Journal of Medical Genetics Part C: Seminars in Medical Genetics . 169 (3): 239–250. doi : 10.1002/ajmg.c.31448 . PMID 26285164 . ^ "Jacobsen Syndrome" . DoveMed . ^ "11q deletion syndrome" . www.socialstyrelsen.se (in Swedish). ^ "Jacobsen syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ^ "Jacobsen syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ^ "11q deletion syndrome" . www.socialstyrelsen.se (in Swedish). ^ "11q deletion syndrome" . www.socialstyrelsen.se (in Swedish). ^ "Jacobsen Syndrome" . Healthline . 2016-11-28. ^ "Jacobsen Syndrome" . prezi.com . ^ "Jacobsen Syndrome" . ... June 2018. ^ "11q deletion syndrome" . www.socialstyrelsen.se (in Swedish). ^ "Jacobsen Syndrome" .FLI1, ETS1, CBL, KIRREL3, UBR1, FLII, FRAXE, JAM3, CCND1, BARX2, TMEM67, CC2D2A, PACC1, AHI1, NTM, RPGRIP1L, NRGN, FZD4, TECTA, NPHP1, NDP, MYH10, MYCN, JBS, IGF1, FRAXA, CTBP2, BSX

-
Craniofacial Abnormality
Wikipedia
External links [ edit ] Classification D ICD - 10 : Q75 ICD - 9-CM : 756.0 MeSH : D019465 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome This human musculoskeletal system article is a stub .TCOF1, SATB2, LMNA, FGF8, MSX2, FGFR2, VSX1, TBX1, SOX9, GTF2IRD1, GPC4, EYA1, TP63, TRPS1, HSPG2, NDST1, DLX5, PDGFRA, LTBP3, SIX1, MSX1, RUNX2, SKI, PRKRA, UFD1, ZIC3, ALX1, FZD4, H4C3, PITX2, MASP1, MBTPS1, PTCH1, FADD, PHEX, UBE3A, TGFBR2, THRB, ACTG1, PTEN, TGFB2, TFAP2B, TFAP2A, RAD23B, TCF4, TBX15, RYK, SIM2, TBCE, SURF1, SRP68, ITGB1BP1, KIAA0586, NRXN1, CHST14, SALL4, ALX4, STRA6, COLEC11, UXS1, PBX4, ZIC5, FOXP2, RAB39B, BNC2, GSC, UNC45B, CNTN4, RDH10, RAB40AL, KANSL1, SH3PXD2B, FAM83H, ANKH, TMCO1, PAX3, MSL3, GPC6, ATP6AP2, DCAF7, SPRY2, IRX5, ZMPSTE24, DLL3, RAI1, KIF3A, RAX, B4GALT7, SLC35D1, NIPBL, SIN3A, SETBP1, B3GAT3, SALL3, SEC61A1, PAX6, ACTB, EDNRA, ERF, ECE1, LPAR1, EDN1, NOTCH1, EGFR, EP300, ERCC6, ETS2, RCAN1, FGD1, FGF3, FGFR1, FOXI1, FOXC2, FMR1, FOLR1, SLC26A2, DNMT3B, GATA2, COL2A1, ANXA1, APAF1, RHOA, ATRX, BMPR1A, BMPR1B, CECR, COL11A1, DNMT3A, COL11A2, HAPLN1, CSNK1A1, CTNNB1, DLX1, DLX2, DLX6, GABRB3, HYDIN2, IDH2, LRP2, IRF6, MMP2, JAG2, MMP13, HOXB1, HOXA3, HOXA1, LETM1, HMX1, MECP2, TRIM37, MNT, GNAQ, SMAD4, SMAD2, SMAD3, GP1BB, TP53, PHF21A, NHLH1, SETD1B, MN1, CSGALNACT1, RIT1, CRY2, TMEM165, GPATCH3, CHD8, CYP2C9, CYP26B1, RPE65, ZNF462, GREB1L, UVRAG, PTH, ASXL3, MIR873, MIR876, PSS, GLRA4, ACAN, JAG1, ALX3, LINGO2, ESCO2, NFIA, BCL2, FBLN7, SP7, PKD2, TASP1, TMEM87B, KDM2B, CHD7, ANKRD11, CHDH, CDKAL1, EPHX1, ETS1, TGFA, EIF4A3, HMGCS1, HLA-A, HCFC1, CCN6, FLI1, HYAL2, CHRD, DCHS1, GTF2I, TWIST1, BARX2, AXIN2, GLI2, HPD, EFNB1, CKAP4, INTS1, DHCR7, DLG1, SHH, GLI3, DCPS, AGO2, SOX4, MMACHC, TBCA, POLR1A, DVL2, MED13L, SPECC1L, DYRK1A, SCFD1, IDUA, PAPPA

-
Mohr–tranebjærg Syndrome
Wikipedia
Unsourced material may be challenged and removed. Find sources: "Mohr–Tranebjærg syndrome" – news · newspapers · books · scholar · JSTOR ( March 2020 ) ( Learn how and when to remove this template message ) Mohr–Tranebjærg syndrome Other names Deafness–dystonia–optic neuronopathy syndrome, Deafness–dystonia–optic atrophy syndrome, Deafness syndrome, progressive, with blindness, dystonia, fractures, and mental deficiency Mohr–Tranebjærg syndrome is inherited in an X-linked recessive manner Mohr–Tranebjærg syndrome ( MTS ) is a rare X-linked recessive syndrome also known as deafness–dystonia syndrome and caused by mutation in the TIMM8A gene. ... Differential diagnosis [ edit ] This is long and includes Arts syndrome Autosomal recessive nonsyndromic sensorineural deafness type DFNB Friedreich ataxia MELAS syndrome Mitochondrial DNA depletion syndrome (encephalomyopathic form with methylmalonic aciduria) McLeod neuroacanthocytosis syndrome Pendred syndrome Usher syndrome type 1 and 2 Wolfram syndrome X-linked spinocerebellar ataxia type 3 and 4 Treatment [ edit ] Cocheal implants are of dubious value in this condition. ... External links [ edit ] Classification D ICD - 10 : G31.8 OMIM : 304700 MeSH : C535808 External resources Orphanet : 52368 GeneReviews/NCBI/NIH/UW entry on Deafness–Dystonia–Optic Neuronopathy Syndrome MTS — a page at NIH website v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia v t e Mitochondrial diseases Carbohydrate metabolism PCD PDHA Primarily nervous system Leigh disease LHON NARP Myopathies KSS Mitochondrial encephalomyopathy MELAS MERRF PEO No primary system DAD MNGIE Pearson syndrome Chromosomal OPA1 Kjer's optic neuropathy SARS2 HUPRA syndrome TIMM8A Mohr–Tranebjærg syndrome see also mitochondrial proteins This article about an ophthalmic disease is a stub .TIMM8A, TBP, TMEM231, KIAA0586, MSH2, MRC1, MLH1, TMEM67, LRRCC1, CEP290, MLRL, MSH6, RIPK3, MPRIP, ACTB, POTEF, MIR34A, DCAF17, HEMGN, TPI1, VEGFA, WT1, XIST, CXCR4, ARHGEF5, TIMM23, SOCS1, TIMELESS, SLC33A1, ABCG2, SART3, MIR186, PEBP4, DNAJC19, CHEK2, STK38, TBC1D9, FITM2, PRRT2, TP53, PANX1, TIMM8B, HAVCR1, MAP1LC3B, CD274, YTHDF1, SMUG1, SDHD, TNF, FHIT, CUX1, COL11A2, CISH, CHGA, CASP8, CASP3, BTK, BRAF, BGLAP, BCL2, ATM, AQP1, AKR1B1, AKT1, AGTR2, AGTR1, AGT, FDXR, FN1, SLC22A3, GCH1, PARP1, REN, PMS2, PIK3CG, PIK3CD, PIK3CB, PIK3CA, ABCB1, NOTCH2, NOS1, MYH1, MUTYH, KRAS, KCNK2, ITK, GYPA, GRM5, PGR-AS1

-
Vici Syndrome
Wikipedia
Vici syndrome Other names Dionisi–Vici–Sabetta–Gambarara syndrome Vici syndrome has an autosomal recessive pattern of inheritance. ... Differential diagnosis [ edit ] This includes ataxia–telangiectasia , Chédiak–Higashi syndrome , DiGeorge syndrome , Griscelli syndrome and Marinesco–Sjögren syndrome . ... Tube feeding may be needed. Eponym [ edit ] Vici syndrome was first described by Carlo Dionisi-Vici et al. ... (April 2002). "Sister and brother with Vici syndrome: Agenesis of the corpus callosum, albinism, and recurrent infections". ... Nature genetics 45(1): 83-87. ^ J Pediatr Ophthalmol Strabismus . 2014 July 1;51(4):214–20 External links [ edit ] Classification D ICD - 10 : Q87.8 OMIM : 242840 MeSH : C535566 External resources Orphanet : 1493 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome

-
Mody 4
Wikipedia
References [ edit ] External links [ edit ] Classification D OMIM : 606392 MeSH : C563451 v t e Disease of the pancreas and glucose metabolism Diabetes Types type 1 type 2 gestational MODY 1 2 3 4 5 6 Complications See Template:Diabetes Abnormal blood glucose levels Hyperglycaemia Oxyhyperglycemia Hypoglycaemia Whipple's triad Insulin disorders Insulin resistance Hyperinsulinism Rabson–Mendenhall syndrome Other pancreatic disorders Insulinoma Insulitis v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions ) [1] This article about an endocrine, nutritional, or metabolic disease is a stub .
-
Mody 6
Wikipedia
External links [ edit ] Classification D OMIM : 606394 MeSH : C565231 v t e Disease of the pancreas and glucose metabolism Diabetes Types type 1 type 2 gestational MODY 1 2 3 4 5 6 Complications See Template:Diabetes Abnormal blood glucose levels Hyperglycaemia Oxyhyperglycemia Hypoglycaemia Whipple's triad Insulin disorders Insulin resistance Hyperinsulinism Rabson–Mendenhall syndrome Other pancreatic disorders Insulinoma Insulitis v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions ) This article about an endocrine, nutritional, or metabolic disease is a stub .
-
Greig Cephalopolysyndactyly Syndrome
Wikipedia
For the Florida school district with the acronym GCPS (Gadsden County Public Schools), see Gadsden County School District . Greig cephalopolysyndactyly syndrome Patient with Greig cephalopolysyndactyly syndrome showing hypertelorism and macrocephaly. Greig cephalopolysyndactyly syndrome is a disorder that affects development of the limbs, head, and face. The features of this syndrome are highly variable, ranging from very mild to severe. ... These diseases include: acrocallosal syndrome , carpenter syndrome , and Gorlin syndrome . [1] Management [ edit ] The main treatment is surgery to fix the abnormalities in the limbs, like syndactyly. ... External links [ edit ] Greig cephalopolysyndactyly syndrome at NLM Genetics Home Reference GeneReview/NIH/UW entry on Greig Cephalopolysyndactyly Syndrome Classification D OMIM : 175700 MeSH : C537300 C537300, C537300 DiseasesDB : 31558 SNOMED CT : 32985001 External resources GeneReviews : Greig Cephalopolysyndactyly Syndrome Orphanet : 380 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions )

-
Adrenocorticotropic Hormone Deficiency
Wikipedia
External links [ edit ] Classification D ICD - 10 : E23.0 ICD - 9-CM : 253.2 OMIM : 201400 MeSH : C535668 C535668, C535668 DiseasesDB : 33423 v t e Pituitary disease Hyperpituitarism Anterior Acromegaly Hyperprolactinaemia Pituitary ACTH hypersecretion Posterior SIADH General Nelson's syndrome Hypophysitis Hypopituitarism Anterior Kallmann syndrome Growth hormone deficiency Hypoprolactinemia ACTH deficiency / Secondary adrenal insufficiency GnRH insensitivity FSH insensitivity LH/hCG insensitivity Posterior Neurogenic diabetes insipidus General Empty sella syndrome Pituitary apoplexy Sheehan's syndrome Lymphocytic hypophysitis Pituitary adenoma v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions ) This article about an endocrine, nutritional, or metabolic disease is a stub .
-
Caudal Regression Syndrome
Wikipedia
Caudal regression syndrome Other names Sacral regression sequence, Sacral agenesis Sacral agenesis Specialty Medical genetics Caudal regression syndrome , or sacral agenesis (or hypoplasia of the sacrum ), is a rare birth defect. ... "Antenatal diagnosis of sacral agenesis syndrome in a pregnancy complicated by diabetes mellitus". ... PMID 2180307 . ^ a b Medline Plus. Caudal Regression Syndrome. https://medlineplus.gov/genetics/condition/caudal-regression-syndrome/#frequency ^ Al Kaissi, Ali; Klaushofer, Klaus; Grill, Franz (19 December 2008). ... (March 2002). "Caudal Regression Syndrome in Twin Pregnancy With Type II Diabetes" . ... External links [ edit ] Classification D ICD - 10 : Q89.8 OMIM : 600145 DiseasesDB : 31157 External resources eMedicine : orthoped/618 Orphanet : 3027 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome

-
Renal Cysts And Diabetes Syndrome
Wikipedia
Renal cysts and diabetes syndrome Other names MODY 5 MODY 5 is inherited in an autosomal dominant manner. Renal cysts and diabetes syndrome ( RCAD ), also known as MODY 5 , is a form of maturity onset diabetes of the young . ... References [ edit ] ^ Bingham C, Hattersley AT (November 2004). "Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta" . ... External links [ edit ] Classification D ICD - 10 : E11.2 OMIM : 137920 MeSH : C535520 External resources Orphanet : 93111 v t e Disease of the pancreas and glucose metabolism Diabetes Types type 1 type 2 gestational MODY 1 2 3 4 5 6 Complications See Template:Diabetes Abnormal blood glucose levels Hyperglycaemia Oxyhyperglycemia Hypoglycaemia Whipple's triad Insulin disorders Insulin resistance Hyperinsulinism Rabson–Mendenhall syndrome Other pancreatic disorders Insulinoma Insulitis v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions ) This article about an endocrine, nutritional, or metabolic disease is a stub .
-
Charge Syndrome
Wikipedia
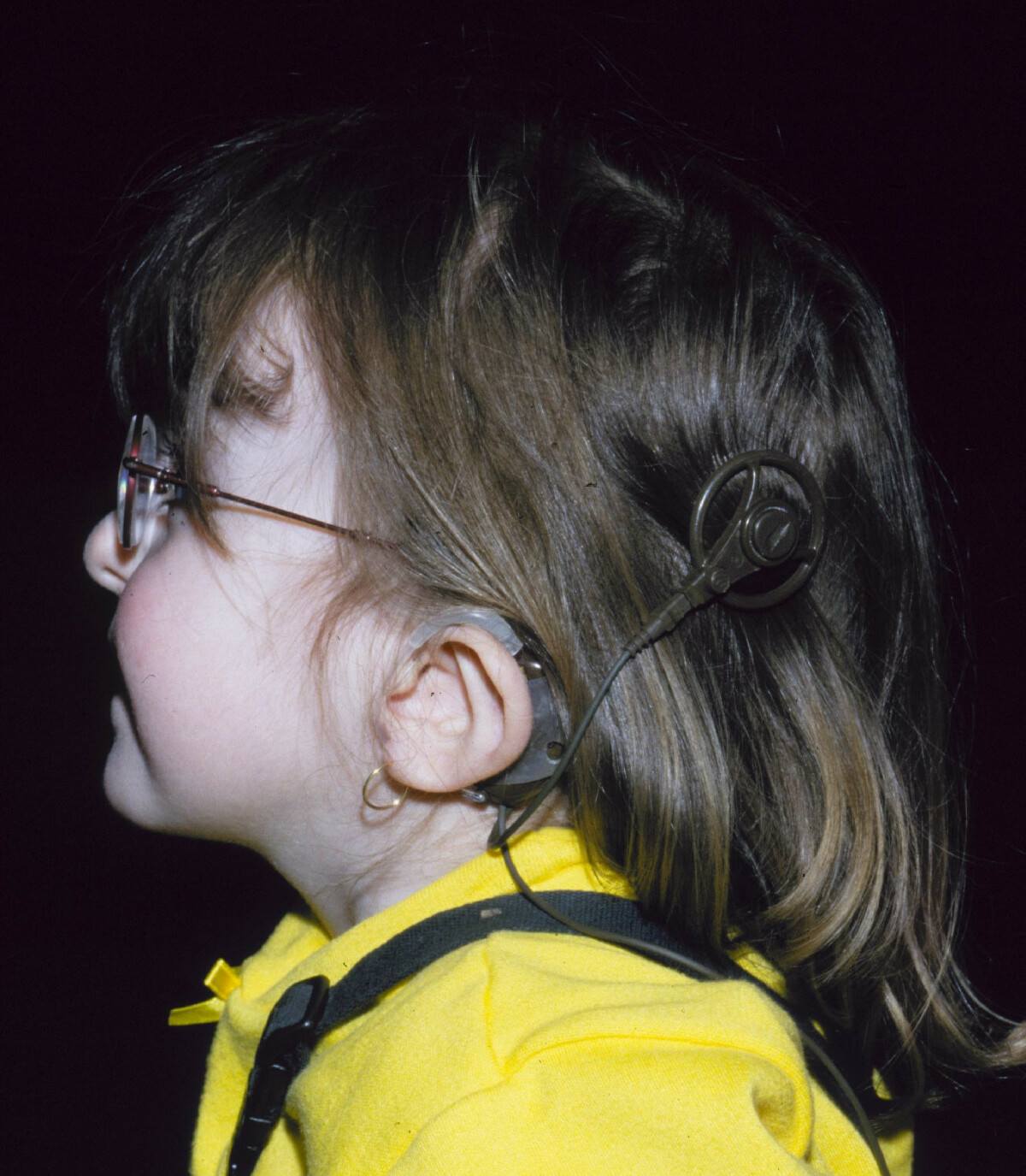
Unsourced material may be challenged and removed. Find sources: "CHARGE syndrome" – news · newspapers · books · scholar · JSTOR ( July 2008 ) ( Learn how and when to remove this template message ) CHARGE syndrome "Lop ear" phenotype characteristic of a person with CHARGE syndrome, along with her cochlear implant . Specialty Medical genetics CHARGE syndrome (formerly known as CHARGE association ) is a rare syndrome caused by a genetic disorder . ... "Spectrum of CHD7 Mutations in 110 Individuals with CHARGE Syndrome and Genotype-Phenotype Correlation" . ... "An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study". ... External links [ edit ] Classification D ICD - 10 : Q87.8 ICD - 9-CM : 759.89 OMIM : 214800 MeSH : D058747 DiseasesDB : 32233 External resources eMedicine : ped/367 v t e Disorders of transcription and post transcriptional modification Chromatin remodeling CHD7 ( CHARGE syndrome ) Polyadenylation PABPN1 ( Oculopharyngeal muscular dystrophy ) RNA splicing PRPF31 ( Retinitis pigmentosa 11 ) PRPF8 ( Retinitis pigmentosa 13 ) v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndromeCHD7, SEMA3E, CHD2, TP53, CHD8, KMT2D, GALNT3, GLUD1, NR0B1, CHDH, ZIC1, FOXN1, MBD2, NRXN1, WASHC5, SEMA3A, SALL4, COA1, FAM124B, FAM172A, TCHP, KDM2B, MTG1, AZIN2, ZNF385B, VIP, SOX2, STATH, RERE, FGF8, FOXE1, FMR1, AFF2, HTC2, IGFALS, IL6, KCNQ2, LEP, CAPRIN1, PAX2, PECAM1, BRD2, SCN2A, SLC12A3, SMARCA1, SMARCA2, SMARCA4, SOD1, GADL1
-
Lymphedema–distichiasis Syndrome
Wikipedia
Lymphedema–distichiasis syndrome Other names Lymphedema with distichiasis [1] Lymphedema–distichiasis syndrome is inherited in an autosomal dominant manner Lymphedema–distichiasis syndrome is a medical condition associated with the FOXC2 gene. [2] : 849 People with this hereditary condition have a double row of eyelashes , which is called distichiasis , and a risk of swollen limbs due to problems in the lymphatic system . ... "Genetic testing for lymphedema-distichiasis syndrome" . The EuroBiotech Journal . 2 (s1): 13–15. doi : 10.2478/ebtj-2018-0026 . ... (eds.), "Lymphedema-Distichiasis Syndrome" , GeneReviews® , University of Washington, Seattle, PMID 20301630 , retrieved 2020-04-15 ^ "Lymphedema-Distichiasis Syndrome" . ... Retrieved 2020-04-15 . ^ Reference, Genetics Home. "Lymphedema-distichiasis syndrome" . Genetics Home Reference . Retrieved 2020-04-15 . ... External links [ edit ] Classification D ICD - 10 : Q82.0 OMIM : 153400 MeSH : C537710 DiseasesDB : 30530 External resources GeneReviews : Lymphedema-Distichiasis Syndrome Orphanet : 33001 GeneReview/NIH/UW entry on Lymphedema-Distichiasis Syndrome v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions )
-
Iridogoniodysgenesis, Dominant Type
Wikipedia
External links [ edit ] Entrez Gene Classification D OMIM : 601631 DiseasesDB : 34611 v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions )
-
Fryns Syndrome
Wikipedia
Fryns syndrome Other names Diaphragmatic hernia-abnormal face-distal limb anomalies syndrome Fryns syndrome is inherited in an autoosomal recessive manner. ... PMID 16440878 . ^ Fryns JP (May 1987). "Fryns syndrome: a variable MCA syndrome with diaphragmatic defects, coarse face, and distal limb hypoplasia" . ... "Terminal deletion of 6q and Fryns syndrome: a microdeletion/syndrome pair?". ... PMID 739533 . ^ Lubinsky M, Severn C, Rapoport JM (March 1983). "Fryns syndrome: a new variable multiple congenital anomaly (MCA) syndrome". ... External links [ edit ] Classification D ICD - 10 : Q87.8 OMIM : 229850 MeSH : C538070 External resources GeneReviews : Fryns syndrome Orphanet : 2059 v t e Congenital abnormality syndromes Craniofacial Acrocephalosyndactylia Apert syndrome Carpenter syndrome Pfeiffer syndrome Saethre–Chotzen syndrome Sakati–Nyhan–Tisdale syndrome Bonnet–Dechaume–Blanc syndrome Other Baller–Gerold syndrome Cyclopia Goldenhar syndrome Möbius syndrome Short stature 1q21.1 deletion syndrome Aarskog–Scott syndrome Cockayne syndrome Cornelia de Lange syndrome Dubowitz syndrome Noonan syndrome Robinow syndrome Silver–Russell syndrome Seckel syndrome Smith–Lemli–Opitz syndrome Snyder–Robinson syndrome Turner syndrome Limbs Adducted thumb syndrome Holt–Oram syndrome Klippel–Trénaunay–Weber syndrome Nail–patella syndrome Rubinstein–Taybi syndrome Gastrulation / mesoderm : Caudal regression syndrome Ectromelia Sirenomelia VACTERL association Overgrowth syndromes Beckwith–Wiedemann syndrome Proteus syndrome Perlman syndrome Sotos syndrome Weaver syndrome Klippel–Trénaunay–Weber syndrome Benign symmetric lipomatosis Bannayan–Riley–Ruvalcaba syndrome Neurofibromatosis type I Laurence–Moon–Bardet–Biedl Bardet–Biedl syndrome Laurence–Moon syndrome Combined/other, known locus 2 ( Feingold syndrome ) 3 ( Zimmermann–Laband syndrome ) 4 / 13 ( Fraser syndrome ) 8 ( Branchio-oto-renal syndrome , CHARGE syndrome ) 12 ( Keutel syndrome , Timothy syndrome ) 15 ( Marfan syndrome ) 19 ( Donohue syndrome ) Multiple Fryns syndrome
-
Focal Dermal Hypoplasia
Wikipedia
These defects manifest as yellow-pink bumps on the skin and pigmentation changes. [2] The disorder is also associated with shortness of stature and some evidence suggests that it can cause epilepsy. [3] Contents 1 Genetics 2 Diagnosis 3 Treatment 4 Eponyms 4.1 Jessner-Cole syndrome 4.2 Goltz-Gorlin 5 See also 6 References 7 External links Genetics [ edit ] The molecular Location of the PORCN gene on the X chromosome: base pairs 48,367,346 to 48,379,201 Focal dermal hypoplasia has been associated with PORCN gene mutations on the X chromosome . [4] 90% of the individuals who are affected with the syndrome are female: the commonly accepted, though unconfirmed, explanation for this is that the non-mosaic hemizygous males are not viable. [5] The differential diagnosis of focal dermal hypoplasia (Goltz) syndrome includes autosomal recessive Setleis syndrome due to TWIST2 gene mutations. It associated with morning glory anomaly, polymicrogyria, incontinentia pigmenti, oculocerebrocutaneous syndrome, Rothmund-Thomson syndrome and microphthalmia with linear skin defects (also known as MLS) syndrome because they are all caused by deletions or point mutations in the HCCS gene. [6] Diagnosis [ edit ] Goltz Syndrome is a very rare diagnosis. To date, there are under 25 cases of Goltz Syndrome in the United States. [7] Treatment [ edit ] Management is targeted toward the various soft tissue and skeletal anomalies, with the goal of achieving optimal functional and cosmetic results. [ citation needed ] Eponyms [ edit ] Jessner-Cole syndrome [ edit ] The disorder was first formally recognized by dermatologists, Max Jessner and Harold Newton Cole, in the early 20th century. ... ISBN 978-0-7216-2921-6 . ^ Goltz RW, Henderson RR, Hitch JM, Ott JE (2008). "Focal dermal hypoplasia syndrome. A review of the literature and report of two cases". ... lng=EN&Expert=2092 GeneReview/NIH/UW entry on Focal dermal hypoplasia Classification D ICD - 10 : Q82.8 ICD - 9-CM : 759.89 OMIM : 305600 MeSH : D005489 DiseasesDB : 29896 External resources eMedicine : derm/155 GeneReviews : Focal Dermal Hypoplasia Orphanet : 2092 v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia










