- Diabetic Embryopathy Wikipedia
-
Postcholecystectomy Syndrome
Wikipedia
. ^ Schmidt M, Søndenaa K, Dumot JA, Rosenblatt S, Hausken T, Ramnefjell M, Njølstad G, Eide GE (28 March 2012). "Post-cholecystectomy symptoms were caused by persistence of a functional gastrointestinal disorder" .
-
Inappropriate Sinus Tachycardia
Wikipedia
"Deciphering the sinus tachycardias" . Clinical Cardiology . 28 (6): 267–76. doi : 10.1002/clc.4960280603 .
-
Meige's Syndrome
Wikipedia
Archived from the original on 2009-02-28. External links [ edit ] Classification D ICD - 10 : G24.4 ICD - 9-CM : 333.82 MeSH : D008538 DiseasesDB : 31428 v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis

- Scleritis Wikipedia
- Fallopian Tube Cancer Wikipedia
-
Sooty Blotch And Flyspeck
Wikipedia
Their 1 month old DNA was extracted and two regions, ITS1 and 28S ribosomal RNA sequenced. Parsimony analysis , bootstrapping and the minimum evolution principle led to groups of species, further described by conidial and colony morphology.
-
Dizziness
Wikipedia
Restorative Neurology and Neuroscience . 28 (1): 83–90. doi : 10.3233/RNN-2010-0530 .ABCB1, CYP2D6, ACHE, ARHGEF2, PFN2, RAP1A, S100B, CCL2, SGSH, SHBG, SRY, TPM3, PER2, CHRNA6, SELENBP1, SPAG9, LGI1, OPRM1, ASIC3, TES, RABGEF1, TERF2IP, CCHCR1, SLC2A4RG, SIL1, EHMT1, PANK2, SPRTN, LRRK2, COPD, GRXCR1, PAEP, NTRK1, ADCY5, FES, APOE, AVP, CHRNA3, CHRNA4, CHRNA7, CHRNB2, CHRNB3, CHRNB4, COMT, CUX1, CYP3A5, DPP4, DSPP, GCG, NT5E, GFAP, GFER, GJA1, GLP1R, GRM5, HINT1, IFNA1, IFNA13, IGF1, IL1B, KCNJ13, MME, NPPA, ZGLP1

-
Blackwater Fever
Wikipedia
"Blackwater fever" (in French). 31 (28). Presse médicale (Paris, France: 1983): 1329–34.
-
Hypertrophic Osteodystrophy
Wikipedia
Veterinary Learning Systems. 23 (9): 22–28. ^ Ettinger, Stephen J.; Feldman, Edward C. (1995).
-
Rubinstein-Taybi Syndrome 2
OMIM
Her mother had preeclampsia during the pregnancy, and the patient was born prematurely at age 28 weeks' gestation. Hamilton et al. (2016) reported heterozygous de novo mutations at highly conserved residues in the EP300 gene in 9 unrelated patients from the UK or Ireland with RSTS2.CREBBP, EP300, SMOC1, EIF4E, PCBP4, PAG1, OPN1LW, MECP2, HTC2, RECQL4, ENOSF1, KAT6B, TWIST1, TNFRSF13B, SRCAP, ZEB2, TMPRSS11D, NIPBL, BMP2, SMARCA4, SMARCA2, SLC20A1, CREB1, POU1F1, OGG1, IGFALS, TNC, HBA2, HBA1, FOXG1, SHH

- Sundowning Wikipedia
-
Fragile X-Associated Tremor/ataxia Syndrome
Wikipedia
.; Rosenwaks, Zev; Yang, Wang-Yong; Gerhardt, Jeannine; Disney, Matthew D.; Jaffrey, Samie R. (2014-02-28). "Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome" .FMR1, C9orf72, FXN, RAN, FMR1-AS1, IGFALS, UBR4, TARDBP, SOD1, IL13, KHDRBS1, MAK16, PLB1, EXOSC7, MIR574, MIR424, SRRM2, NUP62, ATXN10, MIR221, RBMS3, DROSHA, TRA2A, DOCK11, UBQLN2, DCTN4, DGCR8, PNO1, NOP56, DIP2B, IRF2BPL, BEAN1, ASPSCR1, NUFIP2, APOE, PTTG1, PLA2G2A, BRCA2, CDK5, DAXX, FANCD2, MTOR, GRM5, GTF2H1, IGF2R, IL10, LMNA, PLA2G1B, PPP2R2B, SQSTM1, PSEN1, PTBP1, RAD23A, RAD23B, SGCA, SLC1A3, TK2, VIM, YWHAZ, PLA2G6, ATM, HDAC3

-
Hypertelorism
Wikipedia
., PACS: Pediatric Plastic Surgery Volume I, 1984; Chapter 28 Orbital Hypertelorism by Ian T. Jackson ^ Tessier P, Guiot G, Derome P.EFNB1, ACOX1, KAT6B, SPECC1L, SIK3, MED13L, DICER1, GRIP1, TGDS, SUZ12, ATP6V0A2, RPGRIP1L, PIGN, LEMD3, POLR1A, SIN3A, SH2B1, PARS2, SMCHD1, CAMTA1, WDR4, MRAS, TXNL4A, CIT, POLR3A, IL1RAPL1, AP4S1, MAN1B1, CHSY1, KIAA0556, IQSEC2, SPART, NFASC, POGZ, MAPK8IP3, PACS2, NSMF, SETBP1, AUTS2, TCTN3, PSAT1, EFEMP2, VSX1, SLC45A1, SOST, TPRKB, WDPCP, POLR1D, RLIM, TMEM216, RSRC1, WAC, ACTL6B, PTRH2, SUFU, SLC25A24, DSE, BLNK, FOXP1, KIFBP, PHGDH, B3GAT3, FGF20, ELP4, B9D1, AFF4, ANKRD11, INTU, AHDC1, PGAP2, CCDC22, SETD2, UBE2T, CPLX1, ZMYND11, RIPK4, ADAMTS3, DPM1, FGF17, CACNA1G, HERC1, SEMA5A, FIBP, TRIP12, TRIP4, SNAP29, COG1, HS6ST1, RECQL4, EIF2AK3, CHST3, PIGL, CCNK, GPAA1, EED, OFD1, RBM10, SMC1A, LAGE3, TRRAP, LTBP4, OGT, PEX3, PTCH2, CNTNAP1, ITGA8, CDK10, CASK, CDK13, DCHS1, TBX4, POLR1C, RAI1, SEC24C, IRX5, ZMPSTE24, APC2, KLHL41, MAD2L2, ZBTB18, SEC23A, FBLN5, DEAF1, COLEC10, STAMBP, SPINT2, EBP, TBR1, SIX2, LRPPRC, ABCC9, GNE, ZBTB24, TTC37, SEMA3E, TMEM94, KIAA0586, PTDSS1, ZEB2, KIAA0753, AKT3, RUSC2, SEC24D, FIG4, WASHC5, MED12, SLC12A6, FGFRL1, TMCO1, LZTR1, TP53RK, TMEM107, SLX4, KISS1R, MYPN, PIGO, TMEM87B, WDR73, PIGY, COL27A1, UBE3B, RSPRY1, TICRR, TMEM67, STRADA, PGAP3, ANTXR1, KIAA1109, BRIP1, FBXO11, CSPP1, TCTN2, ALG13, GREB1L, FRAS1, CEP290, B9D2, CDCA7, ASXL3, SLC2A10, SPRY4, MED25, DDX59, TRAPPC9, IFT43, CHST14, BNC2, C12orf57, PHACTR1, EBF3, NALCN, KANSL1, PIGW, DOK7, SH3PXD2B, FAM149B1, FREM2, CTU2, KIF7, KBTBD13, GTF2H5, RNU4ATAC, TTN-AS1, JMJD1C, TUBB, TAPT1, B3GLCT, TWIST2, PROKR2, DNAJC21, MPLKIP, AMER1, A2ML1, CCBE1, FLCN, CEP120, ESCO2, FREM1, SPRED1, BMPER, ASXL1, EHMT1, ALG9, PALB2, TBL1XR1, NUP133, NGLY1, ERMARD, MCTP2, CENPJ, HDAC8, KLHL7, SMG9, ALG1, ANKH, LRRC8A, FAM20C, KIF15, CCDC47, KNL1, WDR11, TENM3, VAC14, CEP55, QRICH1, NSUN2, MKS1, PHIP, FANCL, RFWD3, FANCI, PACS1, ASXL2, SLC29A3, CHD7, OSGEP, PIGV, PEX26, RPGRIP1, NUP107, SALL4, BCL11B, FAM111A, MRPS14, XYLT2, NSD1, LMBR1, NXN, TMEM237, BCORL1, CPLANE1, COLEC11, ALG8, TMEM231, FAT4, SRD5A3, PIEZO2, PROK2, THOC2, WDR35, ADGRG6, GATAD2B, TBC1D24, SHROOM4, ARID1B, HACE1, CC2D2A, ALX4, DOCK6, CHD8, ZSWIM6, FANCM, EPG5, KMT2C, USP9X, ALX1, ACTA1, GNRH1, FOXE3, FLI1, FLII, FLNA, FLNB, MTOR, FUCA1, FZD2, GATA6, GBA, GJA1, GK, GPC3, GLB1, GLE1, FOXC1, FH, FGFR2, FBN1, FANCD2, FANCE, BPTF, FANCB, FANCF, FANCG, GPC4, FGFR3, FGD1, FGF3, FGF8, FGF10, FGF14, FGFR1, GLI3, GNRHR, FANCA, GP1BB, KISS1, KRAS, LBR, LETM1, LIG4, LMNA, LOX, LRP2, LRP4, LRP5, SMAD3, SMAD4, MAF, MAT2A, MEF2C, KCNJ2, KCNH1, ANOS1, HNRNPU, GTF2E2, H3-3A, HBA1, HBA2, HELLS, HNRNPH2, HRAS, ITGA3, HSD17B4, HSPG2, IGHM, IGLL1, INPPL1, INSR, FANCC, EZH2, HMGA2, CENPF, BGN, BMP2, BMPR1A, BRCA1, BRAF, BRCA2, BUB1B, CAMK2A, RUNX2, CBL, CCND2, CD79A, CD79B, CDC42, CDH1, NKX3-2, AVP, ATRX, JAG1, ACTA2, ACTB, ACTG1, ACY1, ADK, AGA, AKT1, ATP6V1E1, ALX3, ANK1, APC, ARVCF, ATP6V1A, ATP6V1B2, CDH11, CHD3, EXT2, CHD4, DLX4, DNMT3A, DNMT3B, DPH1, SLC26A2, DUSP6, DVL1, DVL3, DYRK1A, MEGF8, ELN, ERCC2, ERCC3, ERCC4, ERF, DHCR7, DDX3X, CTNND2, COL2A1, CHRNA1, CHRND, CHRNG, CNTN1, COL1A1, COL1A2, COL3A1, CTNND1, COL11A1, COL11A2, COMT, COX7B, CSNK2A1, CTBP1, MID1, KMT2A, MLLT1, TAZ, SET, SIM1, SKI, SKIV2L, SMO, SMS, SOS1, SOS2, SOX9, SOX10, STAT3, STIM1, TAC3, TACR3, MAP3K7, SCN1A, RYR1, RREB1, RET, RAD51C, RAF1, RAPSN, RASA2, RB1, DPF2, RIT1, RRAS, RMRP, RPL5, RPL35A, RPS6KA3, RPS7, RPS19, TBX1, TBCD, MMP2, TBCE, UFD1, UMPS, WHCR, NSD2, NELFA, WNT5A, XRCC2, YWHAE, ZIC1, ZIC3, RNF113A, BRPF1, SHOC2, PDHX, MFAP5, UBE2A, TWIST1, HIRA, TFAP2B, TBX2, TBX15, TCF3, TCF12, TCOF1, TFAP2A, TGFB2, TTN, TGFB3, TGFBR1, TGFBR2, THRA, TPM2, TPM3, RAD51, ALDH18A1, PYCR1, PEX5, TRIM37, MUSK, MYH3, MYH11, MYLK, MYOD1, NEB, NF1, NFIX, TONSL, NONO, NOTCH2, NOTCH3, NRAS, ROR2, TRNW, TRNS2, TRNS1, COX3, ALDH6A1, MOCS1, MOCS2, ATP6, COX1, COX2, ND1, TRNQ, ND4, ND5, ND6, TRNF, TRNH, TRNL1, DDR2, NUP88, PAFAH1B1, PRPS1, PPP2R5D, PPP3CA, PRKAR1A, PRKG1, MAP2K1, MAP2K2, MASP1, PPP2CA, PSMD12, PTCH1, PTEN, PTH1R, PTPN11, PEX2, PPP2R1A, PPP1CB, PAX3, PIGA, PAX6, PDE4D, PDE6D, PEPD, PEX1, PEX6, PIK3CA, POR, PIK3R1, PIK3R2, PITX2, PMM2, EXOSC9, POLR2F, SCN1A-AS1

-
Peripheral Neuropathy, Autosomal Recessive, With Or Without Impaired Intellectual Development
OMIM
The patients ranged in age from 3 to 28 years. Initial features included hypotonia and mildly delayed motor development with most patients achieving walking by age 2 years, although 2 unrelated patients achieved walking at age 4 years.
-
Angiodysplasia
Wikipedia
Transarterial embolization in acute colonic bleeding: review of 11 years of experience and long-term results. Int J Colorectal Dis. 2013 Jun;28(6):777-82". Cite journal requires |journal= ( help ) ^ Junquera F, Saperas E, Videla S, Feu F, Vilaseca J, Armengol JR, Bordas JM, Piqué JM, Malagelada JR (2007).
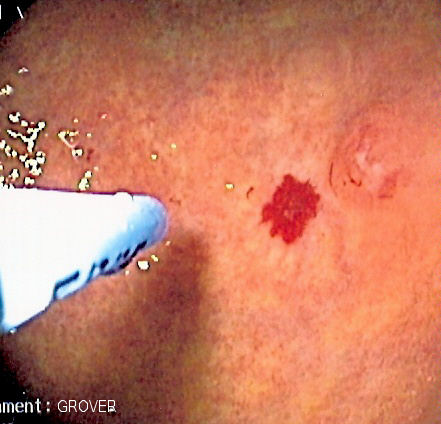
-
Turf Necrotic Ring Spot
Wikipedia
Most infection occurs in spring and fall when the temperature is about 13 to 28°C (5). The primary hosts of this disease are cool-season grasses such as Kentucky bluegrass and annual bluegrass (6).
-
Trichothiodystrophy
Wikipedia
American Journal of Human Genetics . 28 (5): 514–521. PMC 1685097 . PMID 984047 . ^ a b c Freedberg, et al. (2003).ERCC2, ERCC3, MPLKIP, GTF2H5, RNF113A, GTF2E2, GTF2H1, GTF2H3, GTF2H4, GTF2H2, NR1H2, XPC, TP53, HNF1B, HSP90B2P, IFI44, PPARGC1A, SIRT1, XPA, WDR77, DDR1, LBR, SREBF1, MMP1, CAT, ICAM1, HBB, ERCC6, COL6A1, CHEK1, CDK7, CCNH, H3P33

-
Ortner's Syndrome
Wikipedia
Journal of the Saudi Heart Association . 28 (4): 266–269. doi : 10.1016/j.jsha.2016.02.006 .

-
Insensitivity To Pain, Congenital, With Anhidrosis
OMIM
Mapping Shatzky et al. (2000) studied CIPA in consanguineous Israeli-Bedouin groups in which the disorder has a relatively high prevalence. They reported clinical studies of 28 patients. Using the linkage approach, they found that 9 of 10 unrelated families with CIPA were linked to the NTRK1 gene, which had been mapped to chromosome 1q23-q24; in 1 family, linkage was excluded, implying genetic heterogeneity.











