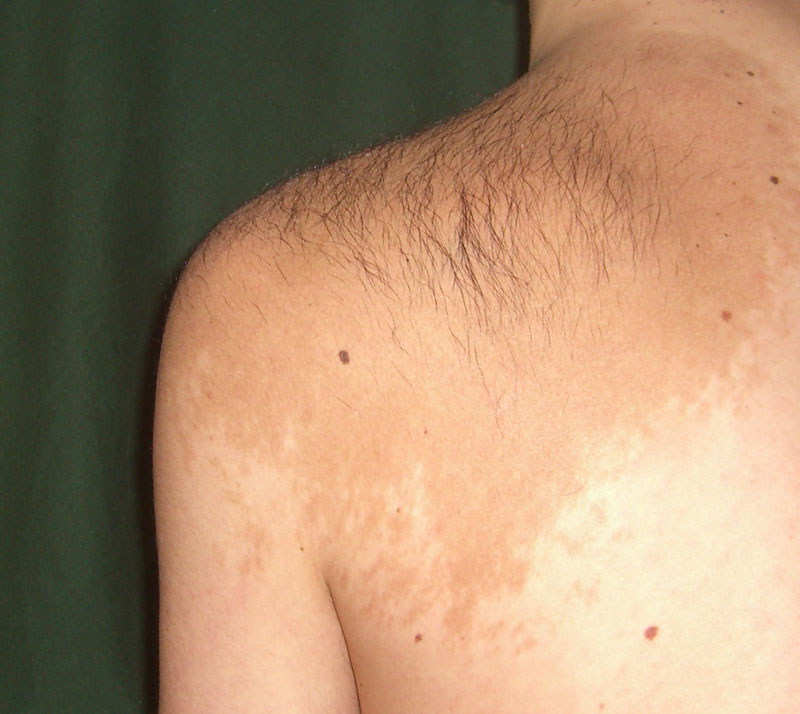
Familial cutaneous telangiectasia and oropharyngeal cancer predisposition syndrome is a rare, inherited cancer-predisposing syndrome characterized by an early development of cutaneous telangiectasia, mild dental and nail anomalies, patchy alopecia over the affected skin areas and increased lifetime risk for oropharyngeal cancer.
A number sign (#) is used with this entry because of evidence that a syndrome involving cutaneous telangiectasia, mild developmental anomalies of hair, teeth, and nails, and a predisposition to cancer, predominantly oropharyngeal, is caused by heterozygous mutation in the ATR gene (601215) on chromosome 3q23. One such family has been reported. Description Patients with this syndrome develop cutaneous telangiectases in infancy with patchy alopecia over areas of affected skin, thinning of the lateral eyebrows, and mild dental and nail anomalies.
Complex regional pain syndrome type 2 (CRPS2), or causalgia is a form of complex regional pain syndrome that develops after damage to a peripheral nerve and is characterized by spontaneous pain, allodynia and hyperalgesia , not necessarily limited to the territory of the injured nerve, as well as at some point, edema, changes in skin blood flow or sudomotor dysfunction in the pain area.
3q27.3 microdeletion syndrome is a rare chromosomal anomaly syndrome, resulting from the partial deletion of the long arm of chromosome 3, characterized by mild to severe intellectual disability, neuropsychiatric disorders of the psychotic and dysthymic spectrum, mild distinctive facial dysmorphism (incl. slender face, deep-set eyes, high nasal bridge with a hooked nose, small, low- set ears, short philtrum, small mouth with thin upper lip, prognathism) and a marfanoid habitus.
Tall stature-scoliosis-macrodactyly of the great toes syndrome is a rare, genetic, overgrowth or tall stature syndrome with skeletal involvement characterized by early and proportional overgrowth, osteopenia, lumbar scoliosis, arachnodactyly of the hands and feet, macrodactyly of the hallux, coxa valga with epiphyseal dysplasia of the femoral capital epiphyses and susceptibility to slipped capital femoral epiphysis.
A number sign (#) is used with this entry because of evidence that the Miura type of epiphyseal chondrodysplasia (ECDM) is caused by heterozygous mutation in the NPR2 gene (108961) on chromosome 9p13. Clinical Features Miura et al. (2012) described a 15-year-old boy, born to nonconsanguineous Japanese parents, with tall stature, scoliosis, long hands and feet with arachnodactyly, and markedly long and broad great toes. His height was greater than 3 SD at birth and at age 15. Bone formation and resorption markers (bone-specific alkaline phosphatase, osteocalcin, cross-linked C-terminal telopeptide of type I collagen, urinary cross-linked N-telopeptide of type I collagen) were elevated. The z score for spine was low (-3.9 corrected for his height). His mother and maternal grandmother had a similar phenotype. Hannema et al. (2013) described a 61-year-old patient with tall stature (+5.2 SD), long fingers, mild scoliosis, and broad but not elongated great toes.
Camptodactyly-taurinuria syndrome is a congenital malformation syndrome characterized by the association of a permanent camptodactyly of the fingers (see this term) with the over excretion of taurine in the urine.
Edinburgh malformation syndrome is a rare, genetic, lethal, multiple congenital anomalies/dysmorphic syndrome characterized by consistently abnormal facial appearance, true or apparent hydrocephalus, motor and cognitive developmental delay, failure to thrive (feeding difficulties, vomiting, chest infections) and death within a few months of birth.
Epilepsy-microcephaly-skeletal dysplasia syndrome is characterized by the association of moderate to severe intellectual deficit, microcephaly, epilepsy, coarse face, hirsutism and skeletal abnormalities (scoliosis and retarded bone development). It has been described only once, in two sibs (one male and one female). This syndrome is likely to be an autosomal recessive condition and thus parents should be informed of a 25% risk of recurrence for other children.
Battaglia et al. (1996) considered this complex to be a new autosomal recessive multiple congenital anomalies/mental retardation syndrome different from Coffin-Lowry syndrome (303600).
Thumb deformity-alopecia-pigmentation anomaly syndrome is a rare, genetic, congenital limb malformation syndrome characterized by short stature, sparse scalp hair, hypoplastic, proximally-placed thumbs, and skin hyperpigmentation with areas of 'raindrop' depigmentation.
Chiba and Miura (1977, 1979) described a mother and son with hypoplastic thumbs and alopecia. Short stature was marked in the child and moderate in the mother; both were reported to be mentally retarded. Winter et al. (1988) reported a similar condition in 4 generations of a family. The affected members studied were a mother and 3 sons. A main distinguishing feature was the presence of a single central upper incisor (147250) in the mother and 1 son of the family reported by Winter et al. (1988). Mental retardation and marked short stature were not found in their family.
Mietens syndrome is a very rare syndrome consisting of corneal opacity, nystagmus, strabismus, flexion contracture of the elbows with dislocation of the head of the radius and abnormally short ulnae and radii.
Mietens and Weber (1966) reported a family in which 4 of 6 offspring of unaffected, second-cousin parents had a syndrome consisting of mental retardation, corneal opacity, nystagmus, strabismus, small pinched nose, flexion contracture of the elbows, dislocation of head of radius, abnormally short ulna and radius, and clinodactyly. Martinez-Glez et al. (2006) described 9-year-old female twins with Mietens-Weber syndrome. The patients were born after a normal pregnancy to young and nonconsanguineous parents.
Freire-Maia et al. (1982) described a 'new' syndrome in 2 brothers with nonconsanguineous parents. ... Features not previously seen in the NFDR syndrome included camptodactyly of the fourth fingers, dysplastic phalanges, ankylosis of the tarsal and metatarsal bones, and dysplastic hips.
Tolosa-Hunt syndrome is an ophthalmoplegic syndrome, affecting all age groups, characterized by acute attacks (lasting a few days to a few weeks) of periorbital pain, ipsilateral ocular motor nerve palsies, ptosis, disordered eye movements and blurred vision usually caused by a non-specific inflammatory process in the cavernous sinus and superior orbital fissure.
Heart-hand syndrome type 2 is an extremely rare heart-hand syndrome (see this term) described in two families to date, that is characterized by upper limb malformations (brachytelephalangy type D, hypoplastic deltoids, mild shortening of the fourth and fifth metacarpals in some individuals, skeletal anomalies in the humerus, radius, ulnae, and thenar bones) and cardiac arrhythmias (junctional rhythms and atrial fibrillation).
Carney complex-trismus-pseudocamptodactyly syndrome is a rare genetic heart-hand syndrome characterized by typical manifestations of the Carney complex (spotty pigmentation of the skin, familial cardiac and cutaneous myxomas and endocrinopathy) associated with trismus and distal arthrogryposis (presenting as involuntary contraction of distal and proximal interphalangeal joints of hands evident only on dorsiflexion of wrist and similar lower-limb contractures producing foot deformities).
., 1992), with Carney complex variant and trismus-pseudocamptodactyly syndrome (158300). In 18 affected members of the family, they identified an arg674-to-gln substitution in the MYH8 gene (R674Q; 160741.0001). ... Stratakis et al. (2004) argued that the syndrome described by Veugelers et al. (2004) was not in fact a variant of Carney complex. They stated that among more than 500 patients with the Carney complex in their database, there was none with trismus-pseudocamptodactyly syndrome. In an analysis of patients reported to have trismus-pseudocamptodactyly syndrome, they identified several with freckling. ... Patients with trismus-pseudocamptodactyly syndrome had none of the lesions or endocrine syndromes typical of Carney complex. Trismus-pseudocamptodactyly syndrome with freckling may or may not be associated with familial myxomas and may or may not be caused by a single mutation of the MYH8 gene, but their own data led Stratakis et al. (2004) to conclude that this disorder is distinct from the Carney complex.
Cardiomyopathy-hypotonia-lactic acidosis syndrome is characterised by hypertrophic cardiomyopathy, muscular hypotonia and the presence of lactic acidosis at birth. It has been described in two sisters (both of whom died within the first year of life) from a nonconsanguineous Turkish family. The syndrome is caused by a homozygous point mutation in the exon 3A of the SLC25A3 gene encoding a mitochondrial membrane transporter.
A number sign (#) is used with this entry because mitochondrial phosphate carrier deficiency can be caused by mutation in the SLC25A3 gene (600370). Clinical Features Mayr et al. (2007) described 2 sisters, offspring of nonconsanguineous Turkish parents, with mitochondrial phosphate carrier deficiency. The younger sister presented at age 12 hours with cyanosis and muscular hypotonia that necessitated intensive care treatment. Echocardiography revealed hypertrophic cardiomyopathy with low cardiac output. Lactate was constantly elevated in plasma. Cardiac hypertrophy was progressive.
A rare, syndromic, benign, epidemal nevus syndrome characterized by the association of a Becker nevus (i.e. circumscribed, unilateral, irregularly shaped, hyperpigmented macules, with or without hypertrichosis and/or acneiform lesions, occuring predominantly on the anterior upper trunk or scapular region) with ipsilateral breast hypoplasia or other, typically hypoplastic, skeletal, cutaneous, and/or muscular defects, such as pectoralis major hypoplasia, supernumerary nipples, vertebral defects, scoliosis, limb asymmetry, odontomaxillary hypoplasia and lipoatrophy.
Clinical Features Happle and Koopman (1997) reviewed 23 cases of a syndrome characterized by the presence of a Becker nevus in association with unilateral hypoplasia of breast or other cutaneous, muscular, or skeletal defects, all of which usually involve the same side of the body as the nevus.
Bartsocas-Papas syndrome is a rare, inherited, popliteal pterygium syndrome (see this term) characterized by severe popliteal webbing, microcephaly, a typical face with short palpebral fissures, ankyloblepharon, hypoplastic nose, filiform bands between the jaws and facial clefts, oligosyndactyly, genital abnormalities, and additional ectodermal anomalies (i.e. absent hair, eyebrows, lashes, nails).
A number sign (#) is used with this entry because of evidence that Bartsocas-Papas syndrome (BPS), also known as the lethal type of popliteal pterygium syndrome, is caused by homozygous mutation in the RIPK4 gene (605706) on chromosome 21q22. ... Description Bartsocas-Papas syndrome (lethal popliteal pterygium syndrome) is an autosomal recessive disorder characterized by multiple popliteal pterygia, ankyloblepharon, filiform bands between the jaws, cleft lip and palate, and syndactyly. ... A less severe form of popliteal pterygium syndrome (119500) is caused by mutation in the IRF6 gene (607199). ... Hall (1984) gave a classification of the lethal pterygium syndromes. In addition to the lethal popliteal pterygium syndrome of Bartsocas and Papas, Hall (1984) recognized 3 lethal multiple pterygium syndromes. ... Papadia and Longo (1988) insisted that there are numerous nosologic differences between Bartsocas-Papas syndrome and lethal multiple pterygium syndrome (253290), 'so that confusion between them is not possible.'
Temtamy preaxial brachydactyly syndrome is a rare, genetic dysostosis syndrome characterized by bilateral, symmetrical, preaxial brachydactyly associated with hyperphalangy, motor developmental delay and intellectual disability, growth retardation, sensorineural hearing loss, dental abnormalities (incuding misalignment of teeth, talon cusps, microdontia), and facial dysmorphism that includes plagiocephaly, round face, hypertelorism, malar hypoplasia, malformed ears, microstomia and micro/retrognathia.
A number sign (#) is used with this entry because of evidence that Temtamy preaxial brachydactyly syndrome (TPBS) is caused by homozygous mutation in the CHSY1 gene (608183) on chromosome 15q26. ... Temtamy et al. (1998) pointed out similarities to several other syndromes, but thought the differences sufficient to consider the disorder in this family to be distinct. They noted that talon cusps have been reported in Rubinstein-Taybi syndrome (180849) and in Mohr syndrome (252100). ... Mapping Li et al. (2010) genotyped DNA samples from the consanguineous Egyptian family with Temtamy preaxial brachydactyly syndrome (TPBS) originally described by Temtamy et al. (1998) and another consanguineous Egyptian family with TPBS, and obtained lod scores of 2.37 and 2.51 at chromosomes 1p33 and 15q26, respectively. ... Tian et al. (2010) noted that hemizygous deletions of CHSY1 occur in the chromosome 15q26-qter deletion syndrome (612626), which shares some phenotypic features in common with TPBS.
Meige syndrome is a rare, neurological condition characterized by blepharospasm (abnormal movement of the eyelids); oromandibular dystonia (spasms in the jaw and tongue); and sometimes, cervical dystonia . Symptoms and severity can vary. The exact cause of Meige syndrome is unknown, but researchers suspect that it is due to a combination of genetic and environmental factors.
Not to be confused with Meigs's syndrome or Meige lymphedema . This article has multiple issues. ... Specialty Neurology Meige's syndrome is a type of dystonia . It is also known as Brueghel's syndrome and oral facial dystonia . ... Treatment [ edit ] In some cases Meige's syndrome can be reversed when it is caused by medication. ... ^ a b c Ananth J, Edelmuth E, Dargan B (April 1988). "Meige's Syndrome Associated with Neuroleptic Treatment". ... Paulson, George W. (December 13, 1997). "Meige's Syndrome" . Blepharospasm.org. Archived from the original on 2012-02-06.
Allan-Herndon-Dudley syndrome is a disorder of brain development that causes moderate to severe intellectual disability and problems with movement. ... Although affected males have speech and a limited ability to communicate, they seem to enjoy interaction with others. Allan-Herndon-Dudley syndrome is caused by mutations in the SLC16A2 gene.
Diagnosis Formal diagnostic criteria for Allan-Herndon-Dudley syndrome have not been established. Suggestive Findings Allan-Herndon-Dudley syndrome (AHDS) should be considered in males with the following clinical findings, brain imaging, and thyroid hormone profiles. ... Molecular Genetic Testing Used in Allan-Herndon-Dudley Syndrome View in own window Gene 1 Method Proportion of Probands with a Pathogenic Variant 2 Detectable by Method SLC16A2 Sequence analysis 3, 4 ~85 5 Gene-targeted deletion/duplication analysis 6 ~15% 5, 7 1. ... Prevalence Prevalence of Allan-Herndon-Dudley syndrome (AHDS) is unknown; however, the identification of more than 160 affected individuals in approximately 15 years suggests that the syndrome is more common than previously thought. ... Treatment of Manifestations in Individuals with Allan-Herndon-Dudley Syndrome (AHDS) View in own window Manifestation/ Concern Treatment Considerations/Other DD/ID See DD/ID Management Issues. ... Recommended Surveillance for Individuals with Allan-Herndon-Dudley Syndrome View in own window System/Concern Evaluation Frequency DD/ID Monitor developmental progress & educational needs.
A number sign (#) is used with this entry because Allan-Herndon-Dudley syndrome (AHDS) is caused by mutation in the MCT8 gene (SLC16A2; 300095) on chromosome Xq13. ... On reevaluation of the family, Bundey et al. (1991) concluded that the disorder may represent the Allan-Herndon syndrome. Bialer et al. (1992) restudied a family reported in abstract by Davis et al. (1981). ... The identification by Dumitrescu et al. (2004) and Friesema et al. (2004) of mutations in the SLC16A2 gene, encoding monocarboxylate transporter-8 (MCT8), in males with hypotonia, involuntary movements, and mental retardation made that gene an attractive candidate for the site of the mutation in Allan-Herndon-Dudley syndrome. Schwartz et al. (2005) found that each of 6 large families with Allan-Herndon-Dudley had mutations in MCT8.
An X-linked intellectual disability syndrome with neuromuscular involvement characterized by infantile hypotonia, muscular hypoplasia, spastic paraparesis with dystonic/athetoic movements, and severe cognitive deficiency. ... Differential diagnosis Differential diagnoses include X-linked intellectual disability conditions associated with ataxia, spastic paraplegia or muscle hypoplasia such as X-linked intellectual disability-spastic paraplegia with iron deposits syndrome, X-linked progressive cerebellar ataxia, and spastic paraplegia type 2. Pelizaeus-Merzbacher disease and Snyder-Robinson syndrome should also be considered. Antenatal diagnosis Antenatal diagnosis of a male with AHDS is possible if the mother is a carrier of a specific SLC16A2 mutation.
Allan–Herndon–Dudley syndrome This condition is inherited in an X-linked recessive manner Specialty Medical genetics , neurology , pediatrics Allan–Herndon–Dudley syndrome is a rare X-linked inherited disorder of brain development that causes both moderate to severe intellectual disability and problems with speech and movement. [1] Allan–Herndon–Dudley syndrome, which is named eponymously for William Allan , Florence C. ... Many people with Allan–Herndon–Dudley syndrome are unable to walk independently and become wheelchair -bound by adulthood. [4] Genetics [ edit ] This condition is inherited in an X-linked recessive pattern. ... Increased T3 levels in the blood may be toxic to some organs and contribute to the signs and symptoms of Allan–Herndon–Dudley syndrome. [ citation needed ] Diagnosis [ edit ] This section is empty. ... American Journal of Mental Deficiency . 48 : 325–34. ^ "Allan-Herndon-Dudley syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ... TRIAC treatment of an infant with Allan-Herndon-Dudley Syndrome (AHDS): Effects on iodothyronines in serum and cerebrospinal fluid . 38th Annual Meeting of the European Thyroid Association.
Allan-Herndon-Dudley syndrome is a rare disorder of brain development that causes moderate to severe intellectual disability and problems with movement. ... Most children with Allan-Herndon-Dudley syndrome have weak muscle tone (hypotonia) and underdevelopment of many muscles (muscle hypoplasia). ... As a result, many people with Allan-Herndon-Dudley syndrome are unable to walk independently and become wheelchair-bound by adulthood. Frequency Allan-Herndon-Dudley syndrome appears to be a rare disorder. ... Increased T3 levels in the blood may be toxic to some organs and contribute to the signs and symptoms of Allan-Herndon-Dudley syndrome. Learn more about the gene associated with Allan-Herndon-Dudley syndrome SLC16A2 Inheritance Pattern This condition is inherited in an X-linked recessive pattern .