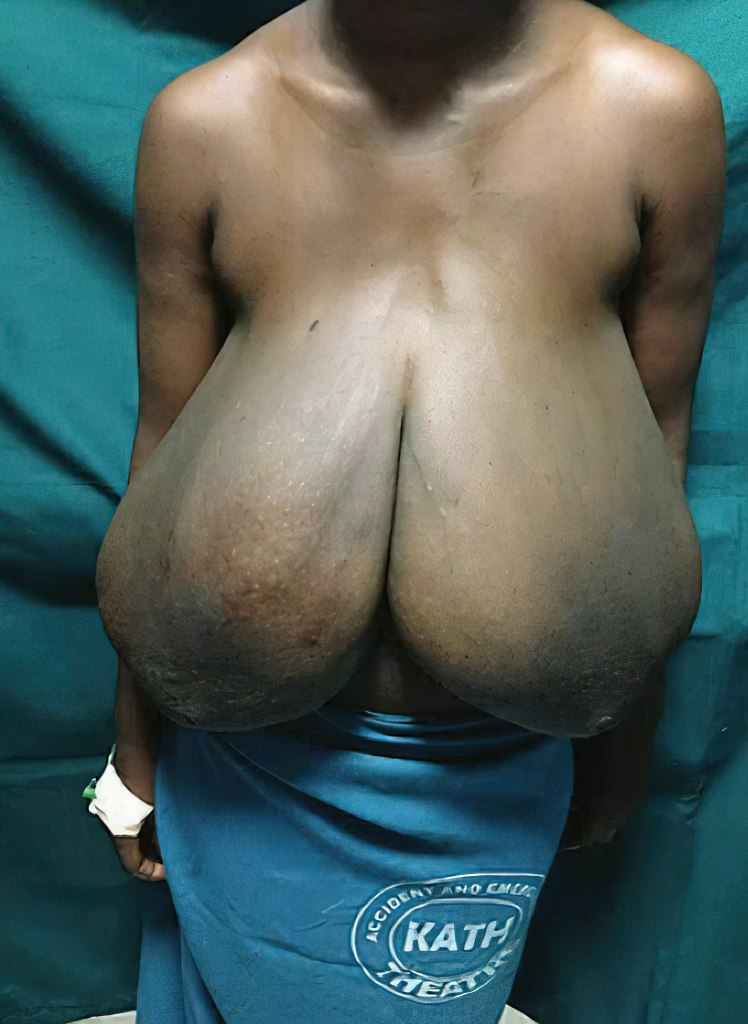
., hepatic growth factor , insulin-like growth factor 1 , and epidermal growth factor ) in the breasts. [23] [24] Macromastic breasts are reported to be composed mainly of adipose and fibrous tissue, while glandular tissue remains essentially stable. [25] Macromastia occurs in approximately half of women with aromatase excess syndrome (a condition of hyperestrogenism ). [26] [27] Hyperprolactinemia has been reported as a cause of some cases of macromastia. [28] [29] Macromastia has also been associated with hypercalcemia (which is thought to be due to excessive production of parathyroid hormone-related protein ) and, rarely, systemic lupus erythematosus [25] and pseudoangiomatous stromal hyperplasia . [30] It is also notable that approximately two-thirds of women with macromastia are obese . [25] Aside from aromatase (as in aromatase excess syndrome), at least two other genetic mutations (one in PTEN ) have been implicated in causing macromastia. [31] [32] A handful of drugs have been associated with gigantomastia, including penicillamine , bucillamine , neothetazone , ciclosporin , indinavir , and prednisolone . [25] [33] [34] Treatment [ edit ] Medical treatment has not proven consistently effective. ... The growth occurred during puberty making it a case of juvenile gigantomastia, but the patient did not seek treatment until the age of 29. [52] Another extreme case was observed on August 28, 2003, when a 24-year-old woman was admitted to the Clinical Center Skopje in Macedonia with gigantomastia of pregnancy and the amount later removed from both breasts was 15 kg (33 lb) in total. [53] [54] A second case in Macedonia was reported when the breasts of a 30-year-old woman from a remote mountain village in eastern Macedonia suddenly grew to more than 30 kilograms (66 lb) total. [54] As the disorder becomes more widely known, media reports have increased.
Women who seek an abortion after the 12-week time limit must apply to a special medical assessment board – called an "abortion board"(Norwegian: ‘abortnemnd’ or ‘primærnemnd’) – that will determine whether or not to grant them an abortion. [28] Scotland: In 2005, 6.1% of abortions were done between 14 and 17 weeks, while 1.6% were performed over 18 weeks. [29] Sweden: In 2005, 5.6% of abortions were carried out between 12 and 17 weeks, and 0.8% at or greater than 18 weeks. [30] Switzerland: In 2016, 10% of abortions performed after the legal term were carried out after week 21 (a total of 36 cases). [31] Of these cases 86% were carried out due to physical problems with the child or mother. [31] United States: In 2003, from data collected in those areas that sufficiently reported gestational age , it was found that 6.2% of abortions were conducted between 13 and 15 weeks, 4.2% between 16 and 20 weeks, and 1.4% at or after 21 weeks. [32] In 2014, the CDC reported that 1.3% of reported abortions (5,578) were performed at 21 weeks of gestation or later. [33] Reasons [ edit ] United States [ edit ] See also: Reasons for abortions Reasons for late terminations of pregnancy include when a pregnant woman's health is at risk or when lethal fetal abnormalities have been detected. [6] [7] A study from 2013 found after excluding abortion "on grounds of fetal anomaly or life endangerment", that women seeking late abortions "fit at least one of five profiles: They were raising children alone, were depressed or using illicit substances, were in conflict with a male partner or experiencing domestic violence, had trouble deciding and then had access problems, or were young and nulliparous". ... Percentage of abortions performed in Scotland by estimated gestation Archived 2007-09-28 at the Wayback Machine . Retrieved May 10, 2007. ^ Nilsson, E., Ollars, B., & Bennis, M.. ... PMID 25437742 . Archived from the original on 28 October 2015 . Retrieved 28 October 2015 .
The outcome was favorable in a majority of the cases, but the infant death rate was 27%. [28] A recent review suggested that COVID-19 appeared to be less lethal to mothers and infants than SARS and MERS but that there may be an increased risk of preterm birth after 28 weeks' gestation. [29] 47 million women in 114 low and middle-income countries are projected by UNFPA to be unable to use modern contraceptives if the average lockdown, or COVID-19-related disruption, continues for 6 months with major disruptions to services: For every 3 months the lockdown continues, assuming high levels of disruption, up to 2 million additional women may be unable to use modern contraceptives.
Without fat, the animal will have a hard time absorbing vitamin D 2 and vitamin D 3 and the lower the fat percentage, the greater the risk of vitamin deficiency, which is true in some athletes who strive to get as lean as possible. [26] Malnutrition [ edit ] Although rickets and osteomalacia are now rare in Britain, osteomalacia outbreaks in some immigrant communities included women with seemingly adequate daylight outdoor exposure wearing typical Western clothing. [27] Having darker skin and reduced exposure to sunshine did not produce rickets unless the diet deviated from a Western omnivore pattern characterized by high intakes of meat, fish, and eggs, and low intakes of high-extraction cereals . [28] [29] [30] In sunny countries where rickets occurs among older toddlers and children, vitamin D deficiency has been attributed to low dietary calcium intakes. ... Ish-Shalom et al. [61] performed a study in elderly women to compare the efficacy and safety of a daily dose of 1500 IU to a weekly dose of 10,500 IU and to a dose of 45,000 IU given every 28 days for two months. They concluded that supplementation with vitamin D can be equally achieved with daily, weekly, or monthly dosing frequencies. ... "Vitamin D: a rapid review". Urologic Nursing (Review). 28 (5): 343–9, 384, quiz 350. PMID 18980100 . ^ van Groningen L, Opdenoordt S, van Sorge A, Telting D, Giesen A, de Boer H (April 2010).
It is also important to note, that deleterious mutation in one of MMR genes alone is not sufficient to cause cancer, but that rather further mutations in other tumour suppressor genes need to occur. [27] Diagnosis [ edit ] A diagnosis of Lynch Syndrome is applied to people with a germline DNA mutation in one of the MMR genes (MLH1, MSH2, MSH6, and PMS2) or the EPCAM gene, identified by genetic testing. [28] Candidates for germline genetic testing can be identified by clinical criteria such as the Amsterdam Clinical Criteria and Bethesda Guidelines, or through tumor analysis by immunohistochemistry (IHC), or microsatellite instability (MSI) testing. [28] Genetic testing is commercially available and consists of a blood test. [ citation needed ] Immunohistochemistry [ edit ] Immunohistochemistry (IHC) is a method that can be used to detect abnormal mismatch repair (MMR) protein expression in tumours that are associated with Lynch syndrome.
Jager et al. (1997) reported studies based on the Danish HNPCC register comprising 28 families that fulfilled the Amsterdam criteria (Vasen et al., 1991): i.e., (1) families should exhibit 3 histologically verified cases of colorectal cancer, of which at least 1 should be diagnosed before the age of 50 years; (2) there should be affected individuals in 2 generations, and 1 of these individuals should be a first-degree relative to the other 2; and (3) familial adenomatous polyposis should be excluded.
Syngal et al. (2000) classified 70 families with suspected hereditary colorectal cancer, excluding familial adenomatous polyposis, by several existing clinical criteria for HNPCC. Of the 70 families, 28 fulfilled the Amsterdam criteria, 39 fulfilled the Modified Amsterdam criteria, 34 fulfilled the Amsterdam II criteria, and 56 fulfilled at least 1 of the 7 Bethesda Guidelines for the identification of HNPCC patients. ... Immunohistochemical analysis revealed 68% (28 of 41) of the HNPCC-related endometrial carcinomas with absent or weak PTEN expression.
A cancer-predisposing condition characterized by the development of colorectal cancer not associated with colorectal polyposis, endometrial cancer, and various other cancers (such as malignant epithelial tumor of ovary, gastric, biliary tract, small bowel, and urinary tract cancer) that are frequently diagnosed at an early age.
A number sign (#) is used with this entry because hereditary nonpolyposis colorectal cancer-4 (HNPCC4) is caused by heterozygous mutation in the PMS2 gene (600259) on chromosome 7p22. Clinical Features Nicolaides et al. (1994) identified a germline deletion in the PMS2 gene in a patient with a family history of HNPCC. A second deletion was found in the patient's tumor sample. The tumor from this patient exhibited microsatellite instability. To examine the contribution of the PMS2 and EXO1 (606063) genes to the HNPCC disease phenotype, Thompson et al. (2004) studied 21 families negative for mutations in MSH2 (609309) and MLH1 (120436) that fulfilled the Amsterdam diagnostic criteria. They found that mutation in PMS2 accounts for only a small proportion of HNPCC families.
A rare inherited cancer-predisposing syndrome characterized by predisposition to a wide variety of cancers, including neoplasms of the digestive tract, urinary tract, kidney, endometrium, ovary, brain, and prostate, as well as sebaceous skin tumors, depending on the gene involved. Tumors may occur at any age but often arise in young people. Factors influencing individual tumor risk include sex, age, affected gene, and personal history of cancer.
A number sign (#) is used with this entry because hereditary nonpolyposis colorectal cancer-7 (HNPCC7) is caused by mutation in the MLH3 gene (604395) on chromosome 14q24.3. For a phenotypic description and a discussion of genetic heterogeneity of hereditary nonpolyposis colorectal cancer, see HNPCC1 (120435). Molecular Genetics Liu et al. (2003) screened index patients from 70 families with colorectal cancer for germline mutations in the MLH3 gene. None of the families had classical or attenuated familial adenomatous polyposis. Liu et al. (2003) identified 1 frameshift mutation and 11 missense mutations in MLH3 in 16 of the 70 index patients (23%).
A number sign (#) is used with this entry because of evidence that hereditary nonpolyposis colorectal cancer-6 is caused by heterozygous mutation in the TGFBR2 gene (190182) on chromosome 3p22. For a phenotypic description and a discussion of genetic heterogeneity of hereditary nonpolyposis colorectal cancer (HNPCC), see HNPCC1 (120435). Clinical Features Among 5 HNPCC families without microsatellite instability, Lu et al. (1998) found a germline missense mutation in the TGFBR2 gene in 1 family. The proband and her 2 brothers had colorectal cancers complying with the clinical criteria A of HNPCC, but the onset of cancer was beyond 50 years of age in all cases (80 in the case of the proband and 65 and 60 in her 2 brothers, respectively), which did not satisfy the Amsterdam criteria. Unlike patients with typical HNPCC, affected members of this family lacked multiple synchronous, metachronous colorectal cancers and extracolonic cancers.
A number sign (#) is used with this entry because hereditary nonpolyposis colorectal cancer-5 (HNPCC5) is caused by heterozygous mutation in the MSH6 gene (600678) on chromosome 2p16. Description Hereditary nonpolyposis colorectal cancer type 5 is a cancer predisposition syndrome characterized by onset of colorectal cancer and/or extracolonic cancers, particularly endometrial cancer, usually in mid-adulthood. The disorder shows autosomal dominant inheritance with incomplete penetrance (summary by Castellsague et al., 2015). For a phenotypic description and a discussion of genetic heterogeneity of hereditary nonpolyposis colorectal cancer (HNPCC), see HNPCC1 (120435). Clinical Features Miyaki et al. (1997) reported a family with HNPCC5.
A number sign (#) is used with this entry because this form of hereditary nonpolyposis colorectal cancer results from heterozygous deletion of 3-prime exons of the EPCAM gene (185535) and intergenic regions directly upstream of the MSH2 gene (609309), resulting in transcriptional read-through and epigenetic silencing of MSH2 in tissues expressing EPCAM. For a phenotypic description and a discussion of genetic heterogeneity of hereditary nonpolyposis colorectal cancer (HNPCC), see HNPCC1 (120435). Molecular Genetics Chan et al. (2006) reported inheritance of germline allele-specific and mosaic hypermethylation of the MSH2 gene (609309), without evidence of DNA mismatch repair gene mutation, in a 3-generation Chinese family. Three sibs carrying the germline methylation developed early-onset colorectal or endometrial cancers, all with microsatellite instability and MSH2 protein loss. Clonal bisulfite sequencing and pyrosequencing showed different methylation levels in different somatic tissues, with the highest level recorded in rectal mucosa and colon cancer tissue, and the lowest in blood leukocytes.
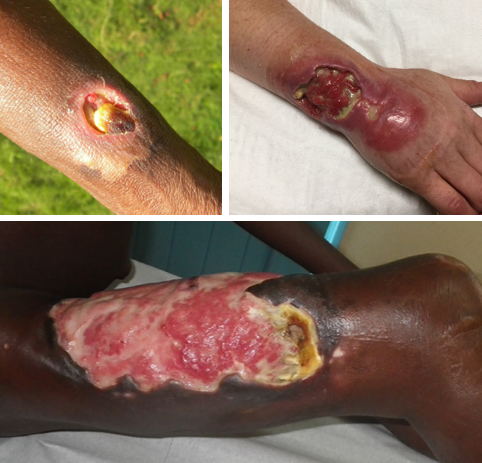
The World Health Organization recommends standard wound care practices: cover the ulcer to keep it moist and protected from further damage; regularly change wound dressings to keep the ulcer clean, remove excess fluid, and help prevent infection. [28] Treatment sometimes includes surgery to speed healing by removing necrotic ulcer tissue, grafting healthy skin over the wound, or removing scar tissue that can deform muscles and joints. [26] [28] Specialized wound dressings developed for non-infectious causes of ulcer are occasionally used for treating Buruli ulcer, but can be prohibitively expensive for low-resource settings. [24] Prevention [ edit ] Buruli ulcer can be prevented by avoiding contact with aquatic environments in endemic areas although this may not be possible for people living in these areas. [24] The risk of acquiring Buruli ulcer can be reduced by wearing long sleeves and pants, using insect repellent, and cleaning and covering any wounds as soon as they are noticed. [10] There is no specific vaccine for preventing Buruli ulcer. [1] The BCG vaccine typically given to children to protect against tuberculosis offers temporary partial protection from Buruli ulcer. [26] [29] Epidemiology [ edit ] Cases of Buruli ulcer reported to the World Health Organization in 2018.
Utilizing this technique it is possible to determine the concentrations of Ap4A in the tears of patients and in such way diagnose objectively if the samples are indicative of dry eye. [26] The tear osmolarity test has been proposed as a test for dry eye disease. [27] Tear osmolarity may be a more sensitive method of diagnosing and grading the severity of dry eye compared to corneal and conjunctival staining, tear break-up time, Schirmer test, and meibomian gland grading. [28] Others have recently questioned the utility of tear osmolarity in monitoring dry eye treatment. [19] Prevention [ edit ] Avoiding refractive surgery (LASIK & PRK), limiting contact lens use, limiting computer screen use, avoiding environmental conditions can decrease symptoms. [29] Complications can be prevented by use of wetting and lubricating drops and ointments. [30] Treatment [ edit ] A variety of approaches can be taken to treatment. ... February 2013. Archived from the original on 28 July 2016 . Retrieved 29 July 2016 . ^ a b c d Kanellopoulos, AJ; Asimellis, G (2016). ... "Effect of long-term immunosuppression in kidney-graft recipients on cancer incidence: randomised comparison of two cyclosporin regimens". The Lancet . 351 (9103): 623–28. doi : 10.1016/S0140-6736(97)08496-1 . ... Merck Veterinary Manual . Retrieved 28 October 2020 . ^ a b c d Gelatt, Kirk N., ed. (1999). ... Further reading [ edit ] Maskin, Steven L. (2007-05-28). Reversing Dry Eye Syndrome: Practical Ways to Improve Your Comfort, Vision, and Appearance .
Cuba declared the disease eliminated in the 1990s, and in 2004 the Centers for Disease Control and Prevention announced that both the congenital and acquired forms of rubella had been eliminated from the United States . [27] [28] The World Health Organisation declared Australia rubella free in October 2018. [29] Screening for rubella susceptibility by history of vaccination or by serology is recommended in the United States for all women of childbearing age at their first preconception counseling visit to reduce incidence of congenital rubella syndrome (CRS). [30] It is recommended that all susceptible non-pregnant women of childbearing age should be offered rubella vaccination. [30] Due to concerns about possible teratogenicity, use of MMR vaccine is not recommended during pregnancy. [30] Instead, susceptible pregnant women should be vaccinated as soon as possible in the postpartum period . [30] In susceptible people passive immunization , in the form of Polyclonal immunoglobulins appears effective up to the fifth day post-exposure. [31] Treatment [ edit ] There is no specific treatment for rubella; however, management is a matter of responding to symptoms to diminish discomfort. ... J Long Term Eff Med Implants . 15 (3): 319–28. doi : 10.1615/JLongTermEffMedImplants.v15.i3.80 .
Overview Rubella is a contagious viral infection best known by its distinctive red rash. It's also called German measles or three-day measles. This infection may cause mild or no symptoms in most people. However, it can cause serious problems for unborn babies whose mothers become infected during pregnancy. Rubella isn't the same as measles, but the two illnesses share some signs and symptoms, such as the red rash. Rubella is caused by a different virus than measles, and rubella isn't as infectious or as severe as measles.
Ultrasonography is sometimes used to evaluate a suspected kidney mass, as it can characterize cystic and solid kidney masses without radiation exposure and at relative low cost. [12] Radiologically tumors are grouped based on appearance into simple cystic, complex cystic, or solid. [12] The most important differentiating feature of a cancerous and non-cancerous tumor on imaging is enhancement. [24] Simple cysts, which are defined by strict criteria [25] are safe to be monitored if the person does not have any symptoms. [12] However, all masses that are not clearly simple cysts should be further evaluated and confirmed by alternate imaging techniques. [26] [12] Computed tomography (CT) of the abdomen administered with and without IV contrast is the ideal imaging to diagnose and stage kidney cancer. [27] [26] [12] There is tentative evidence that iodinated contrast agents may cause worsening of kidney function in people with chronic kidney disease (CKD) with a glomerular filtration rate (GFR) less than 45ml/min/1.73m 2 and should therefore be given cautiously in this group. [28] Abdominal magnetic resonance imaging (MRI) is an alternative imaging method that can be used to characterize and stage a kidney mass. [29] [26] [12] It may be suggested if contrast material cannot be given. [29] MRI can also evaluate the inferior vena cava if the mass is suspected to extend outside the kidney. [29] Since the lungs are the most common organ for kidney cancer to spread to, a chest X-ray or CT scan may be ordered based on the person's risk for metastatic disease. [12] [26] Classification [ edit ] Micrograph of a kidney cancer (chromophobe renal cell carcinoma , oncocytic variant), that may be challenging to differentiate from a benign kidney tumour ( renal oncocytoma ). ... "Prevention of kidney cancer incidence and recurrence: lifestyle, medication and nutrition". Current Opinion in Urology . 28 (1): 62–79. doi : 10.1097/MOU.0000000000000454 .
Kidney tumour Other names Kidney tumors, renal tumours Micrograph of a renal oncocytoma , a type of benign kidney tumour. H&E stain . Specialty Oncology , nephrology Kidney tumours are tumours , or growths, on or in the kidney . These growths can be benign or malignant ( kidney cancer ). Contents 1 Presentation 2 Diagnosis 2.1 Renal ultrasonography 2.2 Classification 2.2.1 Malignant (cancerous) 2.2.2 Benign 2.3 Surgical complexity 3 Epidemiology 4 References 5 External links Presentation [ edit ] Kidney tumours may be discovered on medical imaging incidentally (i.e. an incidentaloma ), or may be present in patients as an abdominal mass or kidney cyst , hematuria , abdominal pain , or manifest first in a paraneoplastic syndrome that seems unrelated to the kidney. [1] Other markers or complications that may arise from kidney tumours can appear to be more subtle, including; low hemoglobin, fatigue, nausea, constipation, and/or hyperglycemia. [2] Diagnosis [ edit ] Unspecific cortical lesion on CT scan is confirmed cystic and benign with contrast-enhanced renal ultrasonography . A CT scan is the first choice modality for workup of solid masses in the kidneys. Nevertheless, hemorrhagic cysts can resemble renal cell carcinomas on CT, but they are easily distinguished with Doppler ultrasonography (Doppler US).
Schizophrenia has been linked to decreased brain zinc levels. [24] Evidence suggests that zinc deficiency could play a role in depression. [24] [25] [26] Zinc supplementation may be an effective treatment in major depression. [27] [28] Growth [ edit ] Zinc deficiency in children can cause delayed growth [5] and has been claimed to be the cause of stunted growth in one third of the world's population. [29] During pregnancy [ edit ] Zinc deficiency during pregnancy can negatively affect both the mother and fetus. ... "Use of serum zinc concentration as an indicator of population zinc status". Food and Nutrition Bulletin . 28 (3 Suppl): S403-29. doi : 10.1177/15648265070283S303 .
KIT receptor tyrosine kinase and phospho - KIT have been suggested to play a role in the development of pediatric ependymomas, [12] NOTCH1 mutations have been found in approximately 8% of pediatric ependymomas, [22] and MEN1 mutations are occasionally found in pediatric ependymomas. [23] MMP2 and MMP14 appear to also play a role in tumor growth and progression in intracranial cases. [24] Two candidate genes, TPR and CHIBBY , have been identified on commonly altered chromosome regions in pediatric ependymomas, chromosomes 1q25 and chromosome 22q12-q13 . [25] Expression of two additional candidate genes , S100A6 and S100A4 on chromosome 1q have also been found to correspond to supratentorial tumor development and tumors occurring before the age of 3 years old, though it is unclear exactly what role these genes play in the etiology . [26] Tumor progression [ edit ] Ependymomas have been suggested to arise from radial glial cells , suggesting neural stem cell maintenance pathways such as Notch , sonic hedgehog (SHH), and p53 are important for the pathogenesis of ependymomas. [1] Notch signaling pathway and HOX family of transcription factors are up regulated in supratentorial and spinal ependymomas respectively. [1] Over-expression of Notch ligands , receptors , and target genes ( HES1 , HEY2 , and MYC ), as well as down-regulation of Notch repressor (Fbxw7) are found in pediatric ependymoma. [22] Inhibition of Notch pathway impairs tumor growth in vitro . [22] Notch target ErbB2 is up-regulated in most ependymomas, correlating with poor outcome. [8] While p53 ( TP53 ) mutations are not often observed in pediatric ependymoma, [27] the p53 pathway is suggested to play a role in radiation therapy resistance [28] and tumor progression, [29] possibly via over-expression of MDM2 . [30] Further, up-regulation of p73 ( TP73 ), a homolog of p53 , and deletion of the p53 pathway gene p14arf / p16 /INK4A ( CDKN2A ) have also been found in pediatric ependymomas. [3] Over-expression of components of SHH pathway such as GLI1 , GLI2 , and STK36 implicates deregulation of the SHH pathway in ependymomas. [3] Moreover, over-expression of SHH targets IGFBP2 , IGFBP3 , and IGFBP5 in ependymoma is also suggestive of a role for SHH and insulin-like growth factor (IGF) signaling in the pathogenesis of pediatric ependymomas. [31] Rate of progression [ edit ] Endothelial cell KIT expression was associated with a young age at diagnosis of pilocytic astrocytoma or ependymoma. [12] Telomerase activity is found in childhood ependymoma. ... "Prognostic factors in childhood intracranial ependymomas: The role of age and tumor location". Pediatric Neurosurgery . 28 (3): 135–42. doi : 10.1159/000028637 .
In these cases, oral medications are used to lower the BP gradually over 24 to 48 hours. [26] In hypertensive emergency, there is evidence of direct damage to one or more organs. [27] [28] The most affected organs include the brain, kidney, heart and lungs, producing symptoms which may include confusion, drowsiness, chest pain and breathlessness. [26] In hypertensive emergency, the blood pressure must be reduced more rapidly to stop ongoing organ damage, [26] however, there is a lack of randomized controlled trial evidence for this approach. [28] Pregnancy [ edit ] Main articles: Gestational hypertension and Pre-eclampsia Hypertension occurs in approximately 8–10% of pregnancies. [23] Two blood pressure measurements six hours apart of greater than 140/90 mm Hg are diagnostic of hypertension in pregnancy. [29] High blood pressure in pregnancy can be classified as pre-existing hypertension, gestational hypertension , or pre-eclampsia . [30] Pre-eclampsia is a serious condition of the second half of pregnancy and following delivery characterised by increased blood pressure and the presence of protein in the urine . [23] It occurs in about 5% of pregnancies and is responsible for approximately 16% of all maternal deaths globally. [23] Pre-eclampsia also doubles the risk of death of the baby around the time of birth . [23] Usually there are no symptoms in pre-eclampsia and it is detected by routine screening. ... (July 2013). "2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC)" . European Heart Journal . 34 (28): 2159–219. doi : 10.1093/eurheartj/eht151 .
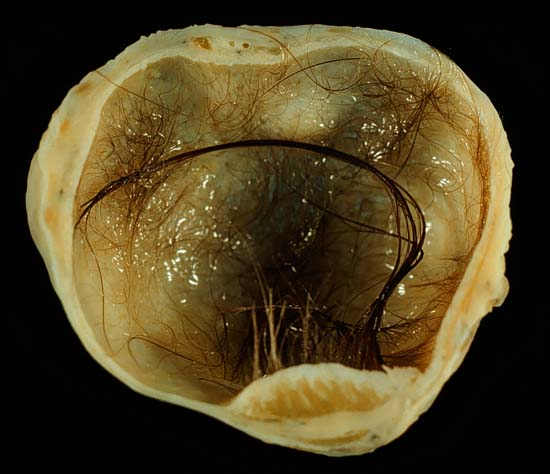
In this condition, the teratomas may contain B cells with NMDA-receptor specificities. [28] After surgery, a risk exists of regrowth in place, or in nearby organs. [29] Pathophysiology [ edit ] Main article: Germ cell tumor Teratomas belong to a class of tumors known as nonseminomatous germ cell tumor . ... "Malignant benign neonatal sacrococcygeal teratoma". J. Pediatr. Surg . 28 (9): 1158–60. doi : 10.1016/0022-3468(93)90154-D .
Extragonadal teratoma is an extremely rare, benign or malignant germ cell tumor characterized, clinically, by a teratoma presenting in an extragonadal location (e.g. retroperitoneum, mediastinum, craniofacial or sacrococcygeal region, intraosseous, solid organs) and, histologically, by displaying well-differentiated structures, as well as immature elements. Presenting symptoms are variable depending on size and location of tumor.
A teratoma with malignant transformation (TMT) is a tumor that develops from germ cells when they grow and divide abnormally, forming a mass . Approximately 6% of teratomas develop into TMTs by a process called malignant transformation , when some of the cells in the teratoma become cancerous. TMTs can occur anywhere in the body, but most are located in the testes in men or ovaries in women. The cause of TMT is unknown. Treatment may include surgery and chemotherapy.
Unsourced material may be challenged and removed. ( February 2016 ) ( Learn how and when to remove this template message ) Genetic Inborn errors of metabolism Congenital disorder of glycosylation [24] Mitochondrial disorders [25] Peroxisomal disorder [26] Glucose transporter defect [27] Menkes disease Congenital disorders of amino acid metabolism [28] Organic acidemia [29] Syndromes Contiguous gene deletion 17p13.3 deletion ( Miller–Dieker syndrome ) [30] Single gene defects Rett syndrome (primarily girls) Nijmegen breakage syndrome X-linked lissencephaly with abnormal genitalia Aicardi–Goutières syndrome Ataxia telangiectasia Cohen syndrome Cockayne syndrome Acquired Disruptive injuries Traumatic brain injury [31] Hypoxic-ischemic encephalopathy [22] Ischemic stroke [21] Hemorrhagic stroke [21] Infections Congenital HIV encephalopathy [32] Meningitis [33] Encephalitis [34] Toxins Chronic kidney failure [35] Deprivation Hypothyroidism [36] Anemia [37] Congenital heart disease [38] Malnutrition [39] Genetic mutations cause most cases of microcephaly. [3] Relationships have been found between autism , duplications of genes and macrocephaly on one side. ... Archived from the original on 2009-05-25. ^ a b c Kalter, Harold (2010-07-28). Teratology in the Twentieth Century Plus Ten .
A rare neurological disorder characterized by a reduced head circumference at birth with no gross anomalies of brain structure. It can be an isolated finding or it can be associated with seizures, developmental delay, intellectual disability, balance disturbances, hearing loss or vision problems.
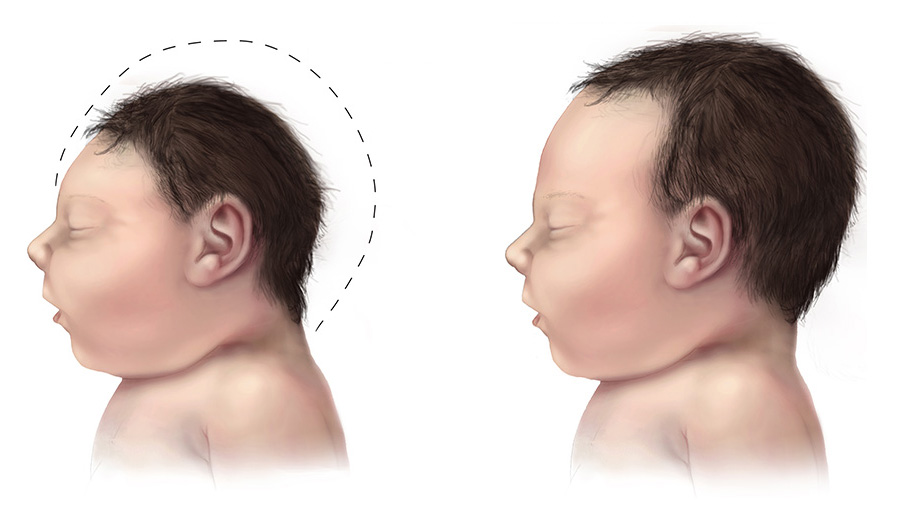
Overview Microcephaly (my-kroh-SEF-uh-lee) is a rare neurological condition in which an infant's head is much smaller than the heads of other children of the same age and sex. Sometimes detected at birth, microcephaly often occurs when there is a problem with brain development in the womb or when the brain stops growing after birth. Microcephaly can be caused by a variety of genetic and environmental factors. Children with microcephaly often have developmental issues. Although there's no treatment for microcephaly, early intervention with speech, occupational and other supportive therapies may help enhance a child's development and improve quality of life. Symptoms The primary symptom of microcephaly is having a head size that is much smaller than that of other children of the same age and sex.
Microcephaly is a rare neurological condition in which a person's head is significantly smaller than expected based on standardized charts. Some cases of microcephaly are detected at birth, while others develop in the first few years of life. Some children with microcephaly have normal intelligence and development. However, microcephaly can be associated with seizures; developmental delay; intellectual disability; problems with movement and balance; feeding difficulties; hearing loss; and/or vision problems depending on the severity of the condition. Because the growth of the skull is determined by brain growth, the condition often occurs when the brain fails to grow at a normal rate.
Elevations in lipase are generally considered a better indicator for pancreatitis as it has greater specificity and has a longer half life. [28] However, both enzymes can be elevated in other disease states. ... Additional tests that may be useful in evaluating chronic pancreatitis include hemoglobin A1C , immunoglobulin G4 , rheumatoid factor , and anti-nuclear antibody . [29] For imaging, abdominal ultrasound is convenient, simple, non-invasive, and inexpensive. [30] It is more sensitive and specific for pancreatitis from gallstones than other imaging modalities. [28] However, in 25–35% of patients the view of the pancreas can be obstructed by bowel gas making it difficult to evaluate. [27] A contrast-enhanced CT scan is usually performed more than 48 hours after the onset of pain to evaluate for pancreatic necrosis and extrapancreatic fluid as well as predict the severity of the disease.
Overview Pancreatitis is inflammation of the pancreas. The pancreas is a long, flat gland that sits tucked behind the stomach in the upper abdomen. The pancreas produces enzymes that help digestion and hormones that help regulate the way your body processes sugar (glucose). Pancreatitis can occur as acute pancreatitis — meaning it appears suddenly and lasts for days. Some people develop chronic pancreatitis, which is pancreatitis that occurs over many years. Mild cases of pancreatitis improve with treatment, but severe cases can cause life-threatening complications.
Incidence of pulmonary adenocarcinoma has been increasing in many developed Western nations in the past few decades, with a share reaching 43.3% of all lung cancers in the US as of 2012, [28] thus replacing squamous cell lung carcinoma as the most common type of lung cancer. ... PMID 25079552 . ^ Mariamidze, Armaz; Aredes, Natália D.; Lee, Jung Il; Rubin, Mark A.; Westervelt, Peter; Tine, Brian Van; Ley, Timothy; Heath, Sharon; Govindan, Ramaswamy (2018-03-28). "Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines" .
Lung adenocarcinoma is a cancer that occurs due to abnormal and uncontrolled cell growth in the lungs. It is a subtype of non-small cell lung cancer that is often diagnosed in an outer area of the lung. Early lung cancers may not be associated with any signs and symptoms. As the condition progresses, affected people can experience chest pain, a persistent cough, fatigue, coughing up blood, loss of appetite, unexplained weight loss, shortness of breath, and/or wheezing. The underlying cause of lung adenocarcinoma is generally unknown; however, risk factors for developing a lung cancer include smoking; exposure to secondhand smoke and other toxic chemicals; a family history of lung cancer; previous radiation treatment to the chest or breast; and HIV infection .
Treatment [ edit ] Main article: Management of hair loss Androgen-dependent [ edit ] Finasteride is a medication of the 5α-reductase inhibitors (5-ARIs) class. [24] By inhibiting type II 5-AR, finasteride prevents the conversion of testosterone to dihydrotestosterone in various tissues including the scalp. [24] [25] Increased hair on the scalp can be seen within three months of starting finasteride treatment and longer-term studies have demonstrated increased hair on the scalp at 24 and 48 months with continued use. [25] Treatment with finasteride more effectively treats male-pattern hair loss at the vertex than male-pattern hair loss at the front of the head and temples. [25] Dutasteride is a medication in the same class as finasteride but inhibits both type I and type II 5-alpha reductase. [25] Dutasteride is approved for the treatment of male-pattern hair loss in Korea and Japan , but not in the United States. [25] However, it is commonly used off-label to treat male-pattern hair loss. [25] Androgen-independent [ edit ] Minoxidil dilates small blood vessels; it is not clear how this causes hair to grow. [26] Other treatments include tretinoin combined with minoxidil, ketoconazole shampoo, dermarolling ( Collagen induction therapy ), spironolactone , [27] alfatradiol , and topilutamide (fluridil). [28] Female pattern [ edit ] There is evidence supporting the use of minoxidil as a safe and effective treatment for female pattern hair loss, and there is no significant difference in efficiency between 2% and 5% formulations. [29] Finasteride was shown to be no more effective than placebo based on low-quality studies. [29] The effectiveness of laser-based therapies is unclear. [29] Procedures [ edit ] More advanced cases may be resistant or unresponsive to medical therapy and require hair transplantation . ... Webmd.com (2010-03-01). Retrieved on 2010-11-28. ^ Rudnicka, L.; Olszewska, M.; Rakowska, A.; Kowalska-Oledzka, E.; Slowinska, M. (2008).
Description Androgenetic alopecia is characterized by a loss of hair from the scalp that follows a defined pattern (Hamilton, 1951). It occurs in women as well as in men. It is caused by a shortening of the anagen (growth) phase and miniaturization of the hair follicle, which results in the formation of progressively thinner, shorter hair (Bergfeld, 1995). In men, the condition is often referred to as male pattern baldness (MPB) and appears to be androgen-dependent (Hamilton, 1942). The condition is hereditary, and follows a pattern that may be consistent with an autosomal dominant trait (Osborn, 1916). Linkage evidence for an autosomal locus on 3q26 (AGA1) has been identified (Hillmer et al., 2008).
Androgenetic alopecia is a common form of hair loss in both men and women. In men, this condition is also known as male-pattern baldness. Hair is lost in a well-defined pattern, beginning above both temples. Over time, the hairline recedes to form a characteristic "M" shape. Hair also thins at the crown (near the top of the head), often progressing to partial or complete baldness. The pattern of hair loss in women differs from male-pattern baldness. In women, the hair becomes thinner all over the head, and the hairline does not recede.
Levinson et al. (1990) reported that, since 1961, 28 families with multiple cases of renal cell carcinoma had been reported, with an abnormality in the constitutional karyotype having been found in only 1 family. ... Anglard et al. (1992) found LOH on 3p in 25 cell lines derived from 28 informative nonpapillary forms of RCC. ... An analysis of patient outcomes showed that 28 of 317 with initially localized disease progressed to metastasis.
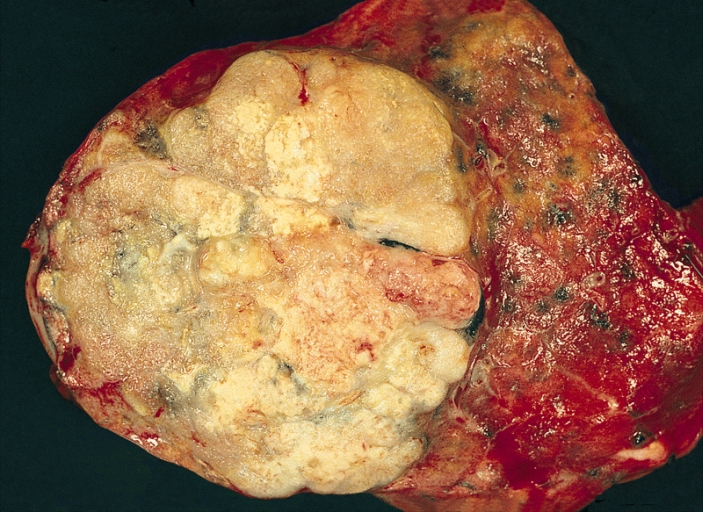
Clear cell renal cell carcinoma is a cancer of the kidney . The name "clear cell" refers to the appearance of the cancer cells when viewed with a microscope.[5258] Clear cell renal cell carcinoma occurs when cells in the kidney quickly increase in number, creating a lump ( mass ). Though the exact cause of clear cell renal cell carcinoma is unknown, smoking, the excessive use of certain medications, and several genetic predisposition conditions (such as von Hippel Lindau syndrome ) may contribute to the development of this type of cancer. Treatment often begins with surgery to remove as much of the cancer as possible, and may be followed by radiation therapy, chemotherapy, biological therapy, or targeted therapy .
A rare renal tumor arising from proximal tubular epithelial cells of the renal cortex, characterized histologically by malignant epithelial cells with typical clear cytoplasm in conventional staining methods due to a high glycogen and lipid content, featuring a nested growth pattern. Clinically it may present with hematuria, flank pain, anemia or, less commonly, a palpable abdominal mass.
The Clear Cell Renal Cell Carcinoma (CCRCC) is a type of renal cell carcinoma . Contents 1 Genetics 1.1 Cytogenetics 1.2 Molecular genetics 2 Histogenesis 3 Microscopy 4 Epidemiology 5 Images 6 References Genetics [ edit ] Cytogenetics [ edit ] Alterations of chromosome 3p segments occurs in 70 – 90% of CCRCCs Inactivation of von Hippel-Lindau disease ( VHL ) gene by gene mutation and promoter hypermethylation Gain of chromosome 5q Loss of chromosomes 8p, 9p, and 14q Molecular genetics [ edit ] Several frequently mutated genes were discovered in CCRCC: VHL , KDM6A /UTX, SETD2 , KDM5C /JARID1C and MLL2 . PBRM1 is also commonly mutated in CCRCC. Histogenesis [ edit ] CCRCC is derived from the proximal convoluted tubule . Microscopy [ edit ] Generally, the cells have a clear cytoplasm, are surrounded by a distinct cell membrane and contain round and uniform nuclei. Microscopically, CCRCCs are graded by the ISUP/WHO as follows: [1] [2] Grade 1: Inconspicuous and basophilic nucleoli at ×400 magnification Grade 2: Clearly visible and eosinophilic nucleoli at ×400 magnification Grade 3: Clearly visible nucleoli at ×100 magnification Grade 4: Extreme pleomorphism or rhabdoid and/or sarcomatoid morphology Epidemiology [ edit ] Most commonly affects male patients in their sixties and seventies.
Hereditary clear cell renal cell carcinoma (ccRCC) is a hereditary renal cancer syndrome defined as development of ccRCC in two or more family members without evidence of constitutional chromosome 3 translocation, von Hippel-Lindau disease or other tumor predisposing syndromes associated with ccRCC, such as tuberous sclerosis or Birt-Hogg-Dubbé syndrome.
Frequency of attacks ranged from 1 per month to 1 per year and typically lasted for several days. The proband was a 28-year-old woman who had migraine with visual aura since age 8 years.
Sporadic hemiplegic migraine is a rare form of migraine headache. Migraines typically cause intense, throbbing pain in one area of the head. Some people with migraines also experience nausea, vomiting, and sensitivity to light and sound. These recurrent headaches typically begin in childhood or adolescence and can be triggered by certain foods, emotional stress, and minor head trauma. Each headache may last from a few hours to a few days. In sporadic hemiplegic migraine and some other types of migraine, a pattern of neurological symptoms called an aura occurs before onset of the headache. An aura commonly includes temporary visual changes such as blind spots (scotomas), flashing lights, zig-zagging lines, and double vision.
Nowaczyk et al. (2000) reported a male infant with ECCL, in whom prenatal sonogram showed normal intracranial structures at 28 weeks of gestation, but at 38 weeks showed asymmetry of the cerebral hemispheres and ventriculomegaly.
A rare, genetic skin disease characterized by the ocular, cutaneous, and central nervous system anomalies. Typical clinical features include a well-demarcated hairless fatty nevus on the scalp, benign ocular tumors, and central nervous system lipomas, leading sometimes to seizures, spasticity, and intellectual disability. Nevus psiloliparus, focal dermal hypo- or aplasia, eyelid skin tags, colobomas, abnormal intracranial vessels, hemispheric atrophy, porencephalic cyst, and hydrocephalus have also been associated.
Encephalocraniocutaneous lipomatosis Other names Haberland syndrome , [1] Specialty Neurology Encephalocraniocutaneous lipomatosis ( ECCL ), is a rare condition primarily affecting the brain , eyes , and skin of the head and face. [2] It is characterized by unilateral subcutaneous and intracranial lipomas , alopecia , unilateral porencephalic cysts , epibulbar choristoma and other ophthalmic abnormalities. It was named after Haberland and Perou who first described it. [3] Contents 1 History 2 See also 3 References 4 External links History [ edit ] This condition was first described in 1970. See also [ edit ] Nevus psiloliparus References [ edit ] ^ Koishi, Giovanna Negrisoli; Yoshida, Mauricio; Alonso, Nivaldo; Matushita, Hamilton; Goldenberg, Dov (2008). "Encephalocraniocutaneous lipomatosis (haberland's syndrome): a case report of a neurocutaneous syndrome and a review of the literature" . Clinics . 63 (3): 406–408. doi : 10.1590/S1807-59322008000300020 . PMC 2664244 .
Encephalocraniocutaneous lipomatosis (ECCL) is a rare condition that primarily affects the brain, eyes, and skin of the head and face. Most of this condition's signs and symptoms are present from birth, and they vary widely among affected individuals. A hallmark feature of ECCL is a noncancerous tumor under the scalp covered by a smooth, hairless patch of skin. This type of tumor, called a nevus psiloliparus, is made up of fatty tissue. Some people with ECCL also have noncancerous tumors under the skin elsewhere on the head or face.