Willems et al. (2008) noted that mutations in the PCNT gene had been identified in 28 patients, including the 25 with MOPD II reported by Rauch et al. (2008) and the 3 diagnosed with Seckel syndrome reported by Griffith et al. (2008).
A rare bone disease and a form of microcephalic primordial dwarfism characterized by severe pre- and postnatal growth retardation, with marked microcephaly in proportion to body size, skeletal dysplasia, abnormal dentition, insulin resistance, and increased risk for cerebrovascular disease. Epidemiology Microcephalic osteodysplastic primordial dwarfism type II (MOPDII) is one of the most common forms of microcephalic primordial dwarfism (MPD) and accounts for more than 150 cases worldwide. Clinical description MOPDII is congenital, with a perinatal and infancy onset. It is characterized by severe pre- and postnatal growth retardation, with proportionate severe microcephaly, skeletal dysplasia, abnormal dentition, an increased risk for cerebrovascular disease (aneurysms and Moya Moya disease in 19%-52% of cases) and insulin resistance. Intrauterine growth restriction (IUGR) is common. The average length, weight, and head occipitofrontal circumference (OFC) at birth are respectively 7.0, 3.9, and 4.6 SDs below the population mean (after correcting for gestational age <37 weeks).
Microcephalic osteodysplastic primordial dwarfism type 2 (MOPD2) is a condition characterized by short stature (dwarfism), skeletal abnormalities and an unusually small head size (microcephaly). Other signs and symptoms of MOPD2 may include hip dysplasia; thinning of the bones in the arms and legs; scoliosis; shortened wrist bones; a high-pitched voice; distinctive facial features (prominent nose, full cheeks, a long midface, and a small jaw); small teeth; abnormal skin pigmentation; and blood vessel abnormalities. Intellectual development is typically normal. It is caused by mutations in the PCNT gene and is inherited in an autosomal recessive manner.
Microcephalic osteodysplastic primordial dwarfism type II (MOPDII) is a condition characterized by short stature (dwarfism) with other skeletal abnormalities (osteodysplasia) and an unusually small head size (microcephaly). The growth problems in MOPDII are primordial, meaning they begin before birth, with affected individuals showing slow prenatal growth (intrauterine growth retardation). After birth, affected individuals continue to grow at a very slow rate. The final adult height of people with this condition ranges from 20 inches to 40 inches. Other skeletal abnormalities in MOPDII include abnormal development of the hip joints (hip dysplasia), thinning of the bones in the arms and legs, an abnormal side-to-side curvature of the spine (scoliosis ), and shortened wrist bones.
Microcephalic osteodysplastic primordial dwarfism type II Other names Majewski osteodysplastic primordial dwarfism type II Microcephalic osteodysplastic primordial dwarfism type II is inherited in an autosomal recessive manner Specialty Medical genetics Microcephalic osteodysplastic primordial dwarfism type II ( MOPD II ) is a form of dwarfism associated with brain and skeletal abnormalities. It was characterized in 1982. [1] MOPD II is listed as a rare disease by the Office of Rare Diseases (ORD) of the National Institutes of Health (NIH). This indicates that MOPD (or a subtype of MOPD) affects less than 200,000 people in the US population. It is associated with the protein pericentrin (PCNT). [2] Notable persons with MOPD II Lucia Zarate , sideshow entertainer See also [ edit ] Primordial dwarfism References [ edit ] ^ Majewski F, Ranke M, Schinzel A (May 1982). "Studies of microcephalic primordial dwarfism II: the osteodysplastic type II of primordial dwarfism".
Esophageal squamous cell carcinoma (ESCC) is a type of esophageal carcinoma (EC; see this term) that can affect any part of the esophagus, but is usually located in the upper or middle third. Epidemiology ESCC has an estimated annual incidence of 1/29,400. Clinical description The average age of onset of ESCC is between the ages of 60 to 70 years and it is more frequently seen in males. It is usually asymptomatic until an advanced disease stage with common presenting symptoms being dysphagia (at first with solids then progressing to fluids) and weight loss. Less commonly odynophagia, hoarseness of voice, coughing, or chest pain can be presenting features. Tumors are typically found in the middle and the upper third of the esophagus.
"Early-Stage, Lymphocyte-Predominant Hodgkin's Lymphoma: Patient Outcomes From a Large, Single-Institution Series With Long Follow-Up". Journal of Clinical Oncology . 1. 28 (1): 136–141. doi : 10.1200/JCO.2009.24.0945 .
Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) is a rare subtype of Hodgkin lymphoma (HL; see this term) characterized histologically by malignant lymphocyte predominant (LP) cells and the absence of typical Hodgkin and Reed-Sternberg (HRS) cells. Epidemiology NLPHL accounts for only 5-10% of HL cases and has an annual incidence of approximately 1/ 830,000. Clinical description Disease onset usually occurs before the age of 40 and there is a 3:1 male predominance for the disease. Unlike classical Hodgkin lymphoma (CHL; see this term) NLPHL has a greater tendency to be restricted to peripheral lymph nodes (neck, axilla or inguino-femoral). Mediastinal involvement is rare and nodal spread is discontiguous. More than 80% of cases present with stage 1 or 2 disease.
Dementia with Lewy bodies is a nervous system disorder characterized by a decline in intellectual function (dementia), a group of movement problems known as parkinsonism, visual hallucinations, sudden changes (fluctuations) in behavior and intellectual ability, and acting out dreams while asleep (REM sleep behavior disorder). This condition typically affects older adults, most often developing between ages 50 and 85. The life expectancy of individuals with dementia with Lewy bodies varies; people typically survive about 5 to 7 years after they are diagnosed. REM sleep behavior disorder may be the first sign of dementia with Lewy bodies. It can occur years before other symptoms appear. Individuals with REM sleep behavior disorder act out their dreams, talking and moving in their sleep when they should be still.
A rare genetic disease characterized by the association of primary lymphedema (typically presenting in one or both lower limbs and frequently affecting the genitalia) and acute myeloid leukemia (often preceded by pancytopenia or myelodysplasia), with or without congenital deafness. Additional reported features include bilateral syndactyly of the toes, hypotelorism and epicanthic folds, long tapering fingers, and neck webbing.
Deafness-lymphedema-leukemia syndrome is a very rare genetic disorder characterized by swelling ( lymphedema ), a weak immune system (immunodeficiency), and blood disorders. Signs and symptoms may include congenital deafness, swelling of the legs and genitalia, a type of cancer known as acute myeloblastic leukemia , reduction of the blood cells (pancitopenia), scarring of the liver (cirrhosis), heart problems and cerebellar atrophy (volume loss of the cerebellum which is part of the brain). Patients may also have closely spaced eyes (hypotelorism), skin folds in the inner corner of the eyes, long tapering fingers and/or neck webbing, recurrent infections in the swelled legs, and many warts. It is caused by a mutation in the GATA2 gene and it is inherited in an autosomal dominant way. Treatment depends on the symptoms.
A number sign (#) is used with this entry because of evidence that primary lymphedema with myelodysplasia is caused by heterozygous mutation in the GATA2 gene (137295) on chromosome 3q21. Immunodeficiency-21 (IMD21; 614172) is an allelic disorder with overlapping clinical features. Clinical Features Emberger et al. (1979) reported a family in which 3 individuals over 2 generations, who had severe congenital deafness, also developed lower limb lymphedema in childhood and hematologic abnormalities, including pancytopenia in 2 of them and acute myeloblastic leukemia in 1 individual. The male proband was born deaf and developed bilateral lymphedema of the lower extremities at 4 years of age. At age 12 years, he was found to have acute myeloblastic leukemia, and died 1 year later.
In a 26-year-old Ashkenazi Jewish man and a 28-year-old Caucasian man with the typical IFAP triad, Oeffner et al. (2011) identified an intronic mutation in the MBTPS2 gene (300294.0007).
X-linked mental retardation, Reish type is characterised by Brain anomalies, severe mental Retardation, Ectodermal dysplasia, Skeletal deformities (vertebral anomalies, scoliosis, polydactyly), Ear/eye anomalies (maldevelopment, small optic nerves, low set and large ears with hearing loss) and Kidney dysplasia/hypoplasia (giving the acronym BRESEK syndrome). Epidemiology It has been described in two brothers, one of whom died shortly after birth. Clinical description One of the brothers also had Hirschsprung disease and Cleft palate/cryptorchidism (giving the acronym: BRESHECK syndrome) Genetic counseling Transmission is X-linked dominant.
This article relies largely or entirely on a single source . Relevant discussion may be found on the talk page . Please help improve this article by introducing citations to additional sources. Find sources: "Ischemic optic neuropathy" – news · newspapers · books · scholar · JSTOR ( May 2011 ) Ischemic optic neuropathy Optic nerve Specialty Ophthalmology Ischemic optic neuropathy ( ION ) is the loss of structure and function of a portion of the optic nerve due to obstruction of blood flow to the nerve (i.e. ischemia ). Ischemic forms of optic neuropathy are typically classified as either anterior ischemic optic neuropathy or posterior ischemic optic neuropathy according to the part of the optic nerve that is affected. People affected will often complain of a loss of visual acuity and a visual field, the latter of which is usually in the superior or inferior field. [1] When ION occurs in patients below the age of 50 years old, other causes should be considered.
. ^ a b c d World Health Organization ICD-10 codes: Diseases of the eye and adnexa (H00-H59). [1] . Retrieved 2010-07-28. ^ International Statistical Classification of Diseases and Related Health Problems. 10th Revision.
Management Entity: Office of International Research, Education, and Development . Archived from the original on 28 August 2009 . Retrieved 29 September 2009 . v t e Culture Outline Sciences Cultural anthropology Cultural astronomy Cultural ecology Cultural geography Cultural neuroscience Cultural studies Culturology Culture theory Security culture Neuroculture Subfields Bioculture Cross-cultural studies Cross-cultural communication Cross-cultural leadership Cross-cultural psychiatry Cross-cultural psychology Cultural analytics Cultural economics Cultural entomology Cultural history Cultural mapping Cultural mediation Cultural psychology Cultural values Culturomics Intercultural learning Intercultural relations Internet culture Philosophy of culture Popular culture studies Postcritique Semiotics of culture Sociology of culture Sound culture Theology of culture Transcultural nursing Types Constructed culture Dominant culture Folk culture High culture Individualistic culture Legal culture Low culture Microculture Official culture Political culture Civic Popular culture Urban Primitive culture Subculture Alternative culture list Super culture Vernacular culture Culture by location Aspects Acculturation Cultural appreciation Cultural appropriation Cultural area Cultural artifact Cultural baggage Cultural behavior Cultural bias Cultural capital Cross-cultural Cultural communication Cultural conflict Cultural cringe Cultural dissonance Cultural emphasis Cultural framework Cultural heritage Cultural icon Cultural identity Cultural industry Cultural invention Cultural landscape Cultural learning Cultural leveling Cultural memory Cultural pluralism Cultural practice Cultural property Cultural reproduction Cultural system Cultural technology Cultural universal Cultureme Enculturation High- and low-context cultures Interculturality Manuscript culture Material culture Non-material culture Organizational culture Print culture Protoculture Safety culture Technoculture Trans-cultural diffusion Transculturation Visual culture Politics Colonial mentality Consumer capitalism Cross cultural sensitivity Cultural assimilation Cultural attaché Cultural backwardness Cultural Bolshevism Cultural conservatism Cultural contracts Cultural deprivation Cultural diplomacy Cultural environmentalism Cultural exception Cultural feminism Cultural genocide Cultural globalization Cultural hegemony Cultural imperialism Cultural intelligence Cultural liberalism Cultural nationalism Cultural pessimism Cultural policy Cultural racism Cultural radicalism Cultural retention Cultural Revolution Cultural rights Cultural safety Cultural silence Cultural subsidy Cultural Zionism Culture change Culture minister Culture of fear Culture war Deculturalization Dominator culture Interculturalism Monoculturalism Multiculturalism Biculturalism Pluriculturalism Polyculturalism Transculturism Religions Buddhism Christianity Catholicism Cultural Catholic Cultural Christian Protestantism Role of Christianity in civilization Eastern Orthodoxy Mormonism Cultural Hindu Islam Cultural Muslim Judaism Cultural Judaism Sikhism Related Animal culture Archaeological culture Bennett scale Cannabis culture Circuit of culture Civilization Coffee culture Cross-cultural Cultural center Cultural competence Cultural critic Cultural determinism Cultural diversity Cultural encoding Cultural evolutionism Cultural homogenization Cultural institution Cultural jet lag Cultural lag Cultural literacy Cultural mosaic Cultural movement Cultural mulatto Cultural probe Cultural relativism Culture speculation Cultural tourism Pop-culture Cultural translation Cultural turn Cultural sensibility Culture and menstruation Culture and positive psychology Culture and social cognition Culture gap Culture hero Culture industry Culture shock Culturgen Children's culture Culturalism Cyberculture Death and culture Disability culture Deaf culture Emotions and culture Intercultural communication Intercultural competence Languaculture Living things in culture Media culture Oppositional culture Participatory culture Permission culture Rape culture Remix culture Tea culture Transformation of culture Urban culture Welfare culture Western culture Category Commons WikiProject Changes Authority control GND : 4226366-9 LCCN : sh85034763
Erdheim-Chester disease (ECD) is a rare condition that can affect many parts of the body. It has been diagnosed in children, but it most commonly affects adults. ECD causes the over-production of immune cells called histiocytes , which then accumulate in tissues and organs in the body. Parts of the body that may be involved include the long bones, retroperitoneum , skin, eyes and eyelids, lungs, brain, heart, kidneys, and pituitary gland; however various other tissues or organs can be affected. The signs and symptoms of ECD vary from person to person depending upon the specific locations and extent of involvement.
Erdheim-Chester disease (ECD), a non-Langerhans form of histiocytosis, is a multisystemic disease characterized by various manifestations such as skeletal involvement with bone pain, exophthalmos, diabetes insipidus, renal impairment and central nervous system (CNS) and/or cardiovascular involvement. Epidemiology Prevalence is unknown. More than 500 cases (<15 pediatric) have been reported since 1930. Clinical description ECD usually presents in adults aged 40-60 with a 3:1 male to female ratio. Clinical course varies from asymptomatic to multisystemic, life-threatening forms. The pathognomonic feature of ECD is osteosclerosis of the long bones manifesting as bone pain, mainly affecting the distal lowerlimbs (50% of cases).
Erdheim-Chester disease is a rare type of slow-growing blood cancer called a histiocytic neoplasm, which results in overproduction of cells called histiocytes. Histiocytes normally function to destroy foreign substances and protect the body from infection. In Erdheim-Chester disease, the excess production of histiocytes (histiocytosis) leads to inflammation that can damage organs and tissues throughout the body, causing them to become thickened, dense, and scarred (fibrotic); this tissue damage may lead to organ failure. People with Erdheim-Chester disease often have bone pain, especially in the lower legs and upper arms, due to an abnormal increase in bone density (osteosclerosis). Damage to the pituitary gland (a structure at the base of the brain that produces several hormones, including a hormone that controls the amount of water released in the urine) may result in hormonal problems such as a condition called diabetes insipidus that leads to excessive urination.
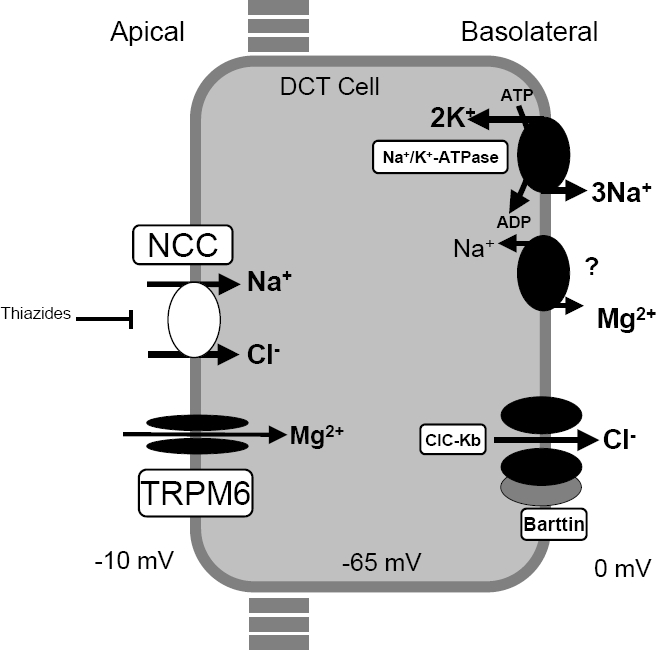
A number sign (#) is used with this entry because of evidence that Gitelman syndrome (GTLMNS) is caused by homozygous or compound heterozygous mutation in the (SLC12A3; 600968) on chromosome 16q13. Description Gitelman syndrome is an autosomal recessive renal tubular salt-wasting disorder characterized by hypokalemic metabolic alkalosis with hypomagnesemia and hypocalciuria. It is the most common renal tubular disorder among Caucasians (prevalence of 1 in 40,000). Most patients have onset of symptoms as adults, but some can present in childhood. Clinical features include transient periods of muscle weakness and tetany, abdominal pains, and chondrocalcinosis (summary by Glaudemans et al., 2012).
A rare syndrome characterized by hypokalemic metabolic alkalosis in combination with significant hypomagnesemia and low urinary calcium excretion. Epidemiology Gitelman syndrome (GS) prevalence is estimated at 1 to 10 per 40,000 and potentially higher in Asia. GS is arguably the most frequent inherited tubulopathy. Clinical description GS presents mainly in adolescents and adults but also encountered in children, as early as in the neonatal period. The diagnosis may be incidental, due to blood tests obtained for unrelated reasons. Clinical symptoms may include salt craving, thirst and nocturia, transient periods of muscle weakness and tetany, sometimes accompanied by abdominal pain.
Gitelman syndrome is a kidney function disorder that causes an imbalance of charged atoms (ions) in the body, including ions of potassium , magnesium , and calcium . It is usually diagnosed during late childhood or adulthood. More common symptoms include fatigue, salt craving, thirst, frequent urination, muscle cramping, muscle weakness, dizziness, tingling or numbness, low blood pressure, and heart palpitations. Gitelman syndrome can be caused by changes (mutations) in the SLC12A3 or CLCNKB genes and is inherited in an autosomal recessive manner. Treatment may include supplementation of potassium and magnesium, and a high sodium and high potassium diet.
Gitelman syndrome is a kidney disorder that causes an imbalance of charged atoms (ions) in the body, including ions of potassium, magnesium, and calcium. The signs and symptoms of Gitelman syndrome usually appear in late childhood or adolescence. Common features of this condition include painful muscle spasms (tetany), muscle weakness or cramping, dizziness, and salt craving. Also common is a tingling or prickly sensation in the skin (paresthesias), most often affecting the face. Some individuals with Gitelman syndrome experience excessive tiredness (fatigue), low blood pressure, and a painful joint condition called chondrocalcinosis.
High Intensity Focused Ultrasound [ edit ] High Intensity Focused Ultrasound (HIFU) is a newer technique for the treatment of malignant and benign tumors of the breast and has shown promising results in the form of complete radiological removal of tumors. [27] An ultrasound beam is focused on a target in the breast and leads to tissue death and protein degradation by raising the temperature in that area. [27] Currently, the use of radiation is recommended in some cases, but HIFU in particular is not part of treatment guidelines. [28] Further research into the usefulness of HIFU, specifically in fibroadenoma, is required before more widespread use of the technique in fibroadenoma. [27] Epidemiology [ edit ] Of all breast tissue samples taken, fibroadenomas comprise about 50%, and this rate rises to 75% for tissue sample in women under the age of 20 years. [29] Fibroadenomas are more frequent among women in higher socioeconomic classes and darker-skinned people. [29] Body mass index and the number of full-term pregnancies were found to have a negative correlation with the risk of fibroadenomas. [29] There are no known genetic factors that influence the rate of fibroadenomas. [29] The rate of occurrence of fibroadenomas in women have been reported in literature to range from 7% to 13%. [29] References [ edit ] ^ 22-251c.Fibroadenomas at Merck Manual of Diagnosis and Therapy Home Edition ^ a b Tavassoli, F.A.; Devilee, P., eds. (2003).
Overview A fibroadenoma (fy-broe-ad-uh-NO-muh) is a solid breast lump. This breast lump is not cancer. A fibroadenoma happens most often between ages 15 and 35. But it can be found at any age in anyone who has periods. A fibroadenoma often causes no pain. It can feel firm, smooth and rubbery. It has a round shape. It might feel like a pea in the breast. Or it may feel flat like a coin. When touched, it moves easily within the breast tissue.
Malignant edema ( or malignant oedema [1] ) is an acute, generally rapidly fatal wound infection ( toxemia ) most common in grazing animals. It affects cattle , horses , sheep , goats , pigs , and deer . It is caused by one or more species of bacteria in the genus Clostridium . [2] [3] "A similar infection in humans is not uncommon." [4] References [ edit ] ^ 'Oedema' is the standard form defined in the Concise Oxford English Dictionary (2011), with the precision that the spelling in the United States is 'edema'. ^ The Merck Veterinary Manual, "Malignant Edema" ^ A World of Petcare, "Malignant Oedema" Archived 2011-07-06 at the Wayback Machine ^ The Merck Veterinary Manual, "Malignant Edema" This veterinary medicine –related article is a stub . You can help Wikipedia by expanding it . v t e
Lung cancer , breast cancer and melanoma patients were found to be at the highest risk of developing brain metastases. [27] [28] [29] [30] [31] However, recent trends in brain metastasis epidemiology have shown an increase in incidence for patients with renal , colorectal or ovarian cancers. [32] Brain metastases are most commonly diagnosed within multiple intracranial areas within the context of extracranial diseases.
Overview Brain metastases occur when cancer cells spread from their original site to the brain. Any cancer can spread to the brain, but the types most likely to cause brain metastases are lung, breast, colon, kidney and melanoma. Brain metastases may form one tumor or many tumors in the brain. As the metastatic brain tumors grow, they create pressure on and change the function of surrounding brain tissue. This causes signs and symptoms, such as headache, personality changes, memory loss and seizures. Treatment for people whose cancer has spread to the brain may include surgery, radiation therapy, chemotherapy, immunotherapy or a combination of treatments.
"Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13". Nature Genetics . 28 (4): 365–370. doi : 10.1038/ng585 .
A number sign (#) is used with this entry because congenital generalized lipodystrophy type 4 (CGL4) is caused by homozygous or compound heterozygous mutation in the PTRF gene (603198) on chromosome 17q21. Description Congenital generalized lipodystrophy type 4 combines the phenotype of classic Berardinelli-Seip lipodystrophy (608594) with muscular dystrophy and cardiac conduction anomalies (Hayashi et al., 2009). For a general description and a discussion of genetic heterogeneity of congenital generalized lipodystrophy, see CGL1 (608594). Clinical Features Rajab et al. (2002) described 10 patients from Oman who had congenital generalized lipodystrophy as well as striking abnormalities in both skeletal and nonskeletal muscle, including reduced exercise tolerance and percussion myoedema. Ghanem (1993) had described percussion myoedema in Berardinelli-Seip lipodystrophy.
A number sign (#) is used with this entry because of evidence that congenital generalized lipodystrophy type 3 (CGL3) is caused by homozygous mutation in the CAV1 gene (601047) on chromosome 7q31. One such family has been reported. Heterozygous mutation in the CAV1 gene can cause familial partial lipodystrophy-7 (FPLD7; 606721). Description Congenital generalized lipodystrophy, also known as Berardinelli-Seip syndrome, is an autosomal recessive disorder characterized by marked paucity of adipose tissue, extreme insulin resistance, hypertriglyceridemia, hepatic steatosis, and early onset of diabetes (Garg, 2004). For a general description and a discussion of genetic heterogeneity of congenital generalized lipodystrophy, see CGL1 (608594). Clinical Features Kim et al. (2008) described a 20-year-old woman, born of consanguineous Brazilian parents, with congenital generalized lipodystrophy.
A number sign (#) is used with this entry because congenital generalized lipodystrophy type 1 (CGL1) is caused by homozygous or compound heterozygous mutation in the gene encoding 1-acylglycerol-3-phosphate O-acyltransferase-2 (AGPAT2; 603100) on chromosome 9q34. Description Congenital generalized lipodystrophy (CGL), or Berardinelli-Seip syndrome, is a rare autosomal recessive disease characterized by a near absence of adipose tissue from birth or early infancy and severe insulin resistance. Other clinical and biologic features include acanthosis nigricans, muscular hypertrophy, hepatomegaly, altered glucose tolerance or diabetes mellitus, and hypertriglyceridemia (Garg, 2004). Genetic Heterogeneity of Congenital Generalized Lipodystrophy Congenital generalized lipodystrophy type 2 (269700) is caused by mutation in the BSCL2 gene (606158). Congenital generalized lipodystrophy type 3 (612526) is caused by mutation in the CAV1 gene (601047).
A number sign (#) is used with this entry because congenital generalized lipodystrophy type 2 (CGL2) is caused by homozygous or compound heterozygous mutation in the gene encoding seipin (BSCL2; 606158) on chromosome 11q12. Biallelic mutation in the BSCL2 gene can also cause progressive encephalopathy with or without lipodystrophy (PELD; 615924), a severe neurodegenerative disorder. Description Congenital generalized lipodystrophy (CGL), also known as Berardinelli-Seip syndrome, is an autosomal recessive disorder characterized by marked paucity of adipose tissue, extreme insulin resistance, hypertriglyceridemia, hepatic steatosis and early onset of diabetes (Garg, 2004). For a general description and a discussion of genetic heterogeneity of congenital generalized lipodystrophy, see CGL1 (608594). Clinical Features Van Maldergem et al. (2002) studied 70 affected individuals from 44 unrelated families with congenital generalized lipodystrophy.
Congenital generalized lipodystrophy is a rare disease characterized by a generalized lack of fat (adipose tissue) in the body. It is part of a group of diseases known as lipodystrophies. Signs and symptoms are noticed from birth (congenital) or early childhood and include high levels of fats (triglycerides) in the blood (hypertriglyceridemia) and insulin resistance (in which the body tissues are unable to respond to the hormone insulin that helps to regulate blood sugar levels) resulting in diabetes mellitus, abnormal accumulation of fat in the liver (liver steatosis) and the accumulation of fat in the heart causing a thickening of the heart muscle ( hypertrophic cardiomyopathy ), which can lead to a heart that does not work well (heart failure) and sudden death. Due to the almost total absence of fatty tissue and excessive growth of muscle tissue, the patients appear very muscular and have visible and prominent veins. They also have dark and thick skin in the body folds (acanthosis nigricans). There are 4 types of the disease that are distinguished by the altered (mutated) genes and by some additional characteristic symptoms.
Summary Clinical characteristics. Berardinelli-Seip congenital lipodystrophy (BSCL) is usually diagnosed at birth or soon thereafter. Because of the absence of functional adipocytes, lipid is stored in other tissues, including muscle and liver. Affected individuals develop insulin resistance and approximately 25%-35% develop diabetes mellitus between ages 15 and 20 years. Hepatomegaly secondary to hepatic steatosis and skeletal muscle hypertrophy occur in all affected individuals. Hypertrophic cardiomyopathy is reported in 20%-25% of affected individuals and is a significant cause of morbidity from cardiac failure and early mortality.
Rare risks of D&E include uterine perforation, retained products of conception, and rare risk of hysterectomy. [14] There is no evidence that surgical abortion causes in increase in infertility or adverse outcomes in subsequent pregnancies. [14] : 252–254 Alternatives [ edit ] Labor induction abortion Medical abortion Complication rates after D&E are similar to or lower than those of labor induction (medical abortion) after 13 weeks, though few studies exist comparing the two approaches. [25] [26] [27] In certain clinical scenarios--severe anemia, for example-- D&E may be preferred over labor induction. [25] Law [ edit ] Proposals to limit abortion access sometimes target specific procedures such as D&E, though this also restricts access for non-abortion patients, such as those with pregnancy loss. [28] See also [ edit ] Late-term abortion References [ edit ] ^ a b "Miscarriage" .
Another proposal in 1993, which was supported by the Archbishop of San Salvador and the "Say Yes to Life Foundation" (an anti-abortion group), would have made December 28, a traditional Roman Catholic feast day known as the Day of the Innocents , the "Day of the Unborn". [2] In 1997, the Nationalist Republican Alliance (ARENA) submitted a draft bill, designed to amend the Penal Code to withdraw all grounds under which abortion was then permitted.