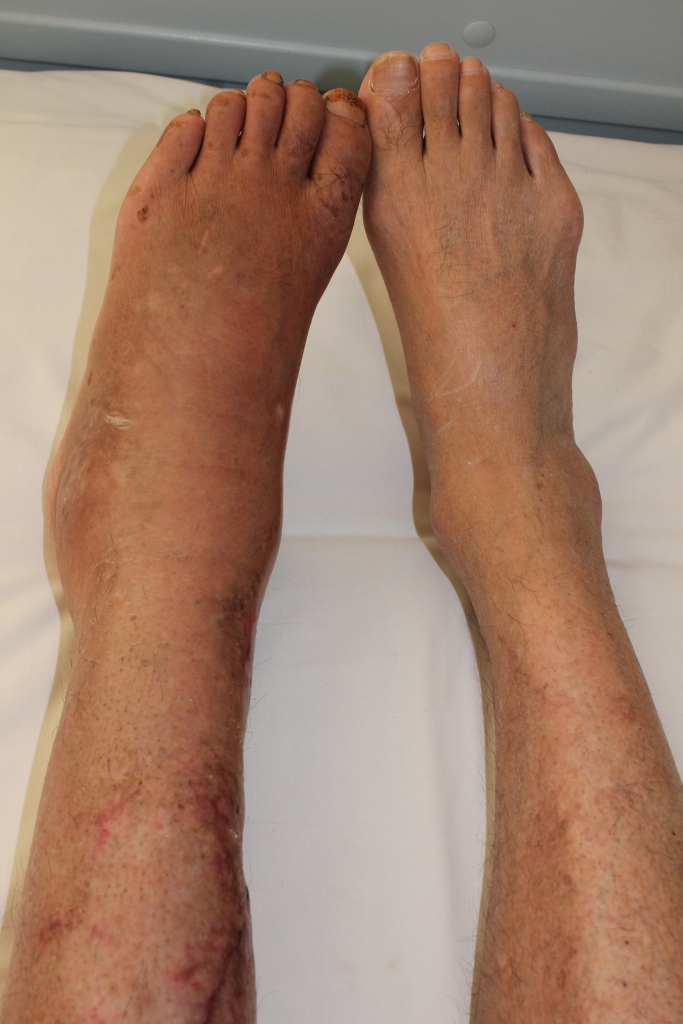
Hemophilia A is the most common form of hemophilia (see this term) characterized by spontaneous or prolonged hemorrhages due to factor VIII deficiency. Epidemiology Prevalence is estimated at around 1 in 6,000 males. Hemophilia primarily affects males, but a symptomatic form of hemophilia A in female carriers (see this term) has also been described with a generally milder clinical picture. Clinical description In general, onset of the bleeding anomalies occurs when affected infants start to learn to walk. The severity of the clinical manifestations depends on the extent of the factor VIII deficiency. If the biological activity of factor VIII is below 1%, the hemophilia is severe and manifests as frequent spontaneous hemorrhage and abnormal bleeding as a result of minor injuries, or following surgery or tooth extraction (severe hemophilia A; see this term).
Moderately severe hemophilia A is a form of hemophilia A (see this term) characterized by factor VIII deficiency leading to abnormal bleeding as a result of minor injuries, or following surgery or tooth extraction. Epidemiology Moderately severe hemophilia A accounts for around 20% of all cases of hemophilia A. Clinical description The biological activity of factor VIII is between 1% and 5%. Spontaneous hemorrhages are rare. Etiology The disorder is caused by mutations in the F8 gene (Xq28) encoding coagulation factor VIII. Genetic counseling Transmission is X-linked recessive.
Hemophilia A is an inherited bleeding disorder in which the blood does not clot normally. People with hemophilia A will bleed more than normal after an injury, surgery, or dental procedure. This disorder can be severe, moderate, or mild. In severe cases, heavy bleeding occurs after minor injury or even when there is no injury (spontaneous bleeding). Bleeding into the joints, muscles, brain, or organs can cause pain and other serious complications. In milder forms, there is no spontaneous bleeding, and the disorder might only be diagnosed after a surgery or serious injury.
Symptomatic hemophilia A in female carriers is a form of hemophilia A (see this term) that manifests in some women with mutations in the F8 gene (Xq28), encoding coagulation factor VIII. Epidemiology Prevalence is unknown but this form of hemophilia is very rare. Clinical description Symptoms include abnormal bleeding as a result of minor injuries, or following surgery or tooth extraction. Spontaneous hemorrhages may occur occasionally. Genetic counseling Transmission is X-linked recessive.
Platelet function disorders include Bernard-Soulier syndrome (OMIM 231200), Glanzmann thrombasthenia (OMIM 273800), and storage pool and nonspecific secretory defects.
Hemophilia is a bleeding disorder that slows the blood clotting process . People with this condition experience prolonged bleeding or oozing following an injury, surgery, or having a tooth pulled. In severe cases of hemophilia, continuous bleeding occurs after minor trauma or even in the absence of injury (spontaneous bleeding). Serious complications can result from bleeding into the joints, muscles, brain, or other internal organs. Milder forms of hemophilia do not necessarily involve spontaneous bleeding, and the condition may not become apparent until abnormal bleeding occurs following surgery or a serious injury.
Endosteal sclerosis-cerebellar hypoplasia syndrome is characterized by congenital cerebellar hypoplasia, endosteal sclerosis, hypotonia, ataxia, mild to moderate developmental delay, short stature, hip dislocation, and tooth eruption disturbances.
Cerebellar hypoplasia with endosteal sclerosis appears to have been described first by Stoll et al. (1986). The parents in this case were consanguineous. Charrow et al. (1991) described a brother and sister and an unrelated boy with congenital cerebellar hypoplasia and endosteal sclerosis. All 3 children presented with ataxia and developmental delay and were found to have microcephaly, short stature, oligodontia, strabismus, nystagmus, and congenital hip dislocation. Ozgen et al. (2005) described the disorder in a girl who they had followed for 11 years. The major clinical symptoms were cerebellar hypoplasia causing ataxia, hypotonia, mild to moderate developmental delay, growth retardation, endosteal sclerosis, tooth eruption disturbances, and hip dislocations.
An extremely rare arthrogryposis syndrome, described in only two pairs of siblings from two unrelated families to date, and characterized by the association of arthrogryposis, congenital torticollis, dysmorphic facial features (i.e. asymmetry of the face, myopathic facial movements, ptosis, posteriorly rotated ears, cleft palate), progressive scoliosis and episodes of malignant hyperthermia.
Froster-Iskenius et al. (1988) believed that this disorder is distinct from the King syndrome (145600) as well as from the multiple pterygium syndrome. Robinson et al. (1987) described a lethal form of multiple pterygium syndrome associated with malignant hyperthermia.
A rare chromosomal anomaly syndrome, resulting from the partial duplication of the long arm of chromosome 22, with a highly variable phenotype principally characterized by developmental delay, intellectual disability, behavioral anomalies, and non-specific craniofacial dysmorphism.
Phocomelia-ectrodactyly-deafness-sinus arrhythmia syndrome is characterised by phocomelia (involving arms more severely), ectrodactyly, ear anomalies (bilateral anomalies of the pinnae), conductive deafness, dysmorphism (long and prominent philtrum, mild maxillary hypoplasia) and sinus arrhythmia.
Stoll et al. (1974) reported father and son with this combination. The arms were severely involved, with absent thumbs and index fingers. The father had bilateral deformity of the pinnae, more marked on the left, with stenosis of the external auditory canal and deafness on that side. The philtrum was long and prominent. The sinus arrhythmia consisted of variable P-P intervals in the electrocardiogram without relationship to respiration. They found no report of this precise combination. Harding et al. (1982) described mother and daughter with radial dysplasia, dysmorphic facies, conductive deafness, and external ear deformity. The facial appearance of the mother suggested mild maxillary hypoplasia.
Taurodontia, absent teeth, sparse hair syndrome is a rare condition that, as the name suggests, is primarily characterized by malformations of the primary and/or secondary molars (taurodontia); the absence of several teeth; and unusually sparse, slow-growing hair.
Clinical Features Stenvik et al. (1972) reported 4 Norwegian sibs with congenitally missing teeth and sparse hair. Three of the 4 sibs had taurodontia. Nails and ability to perspire were not specifically mentioned. The parents were not evaluated. Moller et al. (1973) and Gorlin et al. (1975) reported a female with a similar disorder, the third of 4 children. Sweat function was normal using orthophthalaldialdehyde. The father and one of the proband's sisters congenitally lacked permanent maxillary lateral incisors but were otherwise normal. Gorlin et al. (1975) stated that this combination of findings had previously been reported by Stoy (1960).
Ring chromosome Y syndrome is a rare chromosome Y structural anomaly, with a highly variable phenotype, mostly characterized by short stature, partial to total gonadal failure, sexual infantilism, genital anomalies (e.g. ambiguous genitalia, hypospadias, cryptorchidism), and azoospermia or oligozoospermia.
Complex regional pain syndrome (CRPS) is a rare neurologic disease painful progressive condition that corresponds to a group of disorders characterized by a disproportionate spontaneous or stimulus-induced pain, accompanied by a variably mixed myriad of autonomic and motor disorders including symptoms such as swelling, allodynia, skin blood supply and trophic disturbances.
Complex regional pain syndrome (CRPS) is a chronic pain condition that mainly affects the arms, legs, hands, and feet, but may involve the entire body.
Overview Complex regional pain syndrome (CRPS) is a form of chronic pain that usually affects an arm or a leg. complex regional pain syndrome (CRPS) typically develops after an injury, a surgery, a stroke or a heart attack. ... Diagnosis Diagnosis of complex regional pain syndrome (CRPS) is based on a physical exam and your medical history.
A rare, genetic, congenital limb malformation syndrome characterized by a unique combination of bilateral, symmetrical camptodactyly and clinodactyly of 5th fingers, mesoaxial camptodactyly of toes, and ulnar deviation of 3rd fingers.
Clinical Features Malik et al. (2010) described a 5-generation Pakistani kindred in which 26 members had a unique combination of camptodactyly and clinodactyly of 5th fingers, mesoaxial camptodactyly of toes, ulnar deviation of 3rd fingers, syndactyly involving all digits, and bifid toes. The condition was bilateral and symmetrical and affected upper and lower limbs in all of those affected. The authors noted that syndactyly type II (see 186000) manifests noticeable phenotypic overlap with the clinical presentation in this family, but that the typical syndactyly type II changes such as fusion of fingers 3/4 and toes 2/3 with additional digits in the web were absent; in addition, camptodactyly of mesoaxial toes, ulnar deviation of 3rd fingers, and bifid hallux, which were prominent in this family, are not characteristic of type II syndactyly. This family was originally described by Wahab (1986). Inheritance The pedigree pattern in the family with digit malformations described by Malik et al. (2010) was consistent with autosomal dominant inheritance. There were 4 instances of male-to-male transmission. There was 1 likely case of incomplete penetrance.
Heart defect – round face – congenital developmental delay is very rare syndrome described in three sibs of one Japanese family and characterized by congenital heart disease, round face with depressed nasal bridge, small mouth, short stature, and relatively dark skin and typical dermatoglyphic anomalies, and intellectual deficit.
Clinical Features Among the children of presumably unrelated parents who, however, came from lines that had been located in a rather small town for a long time, Sonoda et al. (1988) described 2 boys and a girl with a syndrome of congenital heart disease, round face with depressed nasal bridge, small mouth, short stature, developmental retardation, relatively dark skin, and high axial triradius in the dermatoglyphic analysis.
A number sign (#) is used with this entry because of evidence that cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS) is caused by a homozygous repeat expansion (AAGGG(n)) in the RFC1 gene (102579) on chromosome 4p14. ... Description Cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS) is an autosomal recessive adult-onset, slowly progressive neurologic disorder characterized by imbalance due to cerebellar gait and limb ataxia, impaired vestibular function bilaterally, and non-length-dependent sensory neuropathy (summary by Szmulewicz et al., 2011). Clinical Features Migliaccio et al. (2004) reported 4 unrelated patients, 2 males and 2 females, with a syndrome comprising cerebellar ataxia and bilateral vestibulopathy with impaired ability of the eye velocity to match head velocity. ... Szmulewicz et al. (2011) reported retrospective data on 27 patients, including the 4 reported by Migliaccio et al. (2004), with a syndrome including cerebellar ataxia, neuropathy, and vestibular areflexia, which they termed 'CANVAS.'
Bent bone dysplasia syndrome is an often lethal skeletal disorder characterized by poor mineralization of the skull (calvarium), craniosynostosis, underdeveloped (hypoplastic) pubic bone (pubis) and clavicles, osteopenia , and bent long bones.
A number sign (#) is used with this entry because of evidence that bent bone dysplasia syndrome (BBDS) is caused by heterozygous mutation in the FGFR2 gene (176943) on chromosome 10q26. ... Molecular Genetics Merrill et al. (2012) analyzed 6 candidate genes in 3 female fetuses and 1 male fetus with a perinatal lethal bent bone dysplasia syndrome and identified heterozygosity for the same de novo missense mutation in the FGFR2 gene in 3 of them (M391R; 176943.0043), with a different heterozygous FGFR2 mutation detected in the remaining fetus (Y381D; 176943.0044).
FGFR2-related bent bone dysplasia is a rare, genetic, lethal, primary bone dysplasia characterized by dysmorphic craniofacial features (low-set, posteriorly rotated ears, hypertelorism, megalophtalmos, flattened and hypoplastic midface, micrognathia), hypomineralization of the calvarium, craniosynostosis, hypoplastic clavicles and pubis, and bent long bones (particularly involving the femora), caused by germline mutations in the FGFR2 gene. Prematurely erupted fetal teeth, osteopenia, hirsutism, clitoromegaly, gingival hyperplasia, and hepatosplenomegaly with extramedullary hematopoesis may also be associated.
A rare developmental defect during embryogenesis syndrome characterized by a glabellar capillary malformation, congenital communicating hydrocephalus, and posterior fossa brain abnormalities, including Dandy-Walker malformation, cerebellar vermis agenesis, and mega cisterna magna.
Dystonia-aphonia syndrome is a rare, genetic, persistent combined dystonia disorder characterized by slowly progressive, severe, caudo-rostrally spreading generalized dystonia with prominent facial and oro-mandibular involvement leading to severe anarthria and/or aphonia, swallowing difficulties, and gait disturbances.
Osteopathia striata-pigmentary dermopathy-white forelock syndrome is characterised by the association of osteopathia striata (longitudinal striations through most of the long bones) with a macular, hyperpigmented dermopathy and a white forelock.
A rare, genetic, multiple congenital anomalies/dysmorphic syndrome characterized by the association of short stature and progressive discrete subaortic stenosis.
From Turkey, Onat et al. (1984) described a family in which the parents were second cousins (according to the pedigree; the authors stated that they were 'offspring of full siblings,' i.e., first cousins) and both had short stature, obstructive lung disease, hoarseness and upturned nose. The father also had aortic stenosis and inguinal hernia; he died suddenly at age 47, having had exertional angina pectoris from age 42. The mother was said to have short, thin, atrophic vocal cords and false cords which assisted phonation. Of 6 offspring, 4 had clinical signs of aortic stenosis. The oldest sib, a woman 147 cm tall, died at age 29 and showed a fibrous ring just below the aortic cusps, together with fibrous thickening of the atrioventricular valves (especially of the mitral valve) and marked shortening of the chordae tendineae. The second sib died at age 23; aortic stenosis had been demonstrated.
An X-linked syndromic intellectual disability characterized by developmental delay, variable degree of intellectual disability, speech delay or absent speech, pyramidal signs, tremor, macroorchidism and variable mood and behavior problems, including psychosis and autistic-like behavior.
Description The MECP2 gene is mutated in Rett syndrome (RTT; 312750), a severe neurodevelopmental disorder that almost always occurs in females. ... Clinical Features Lindsay et al. (1996) described an X-linked mental retardation syndrome linked to Xq28. This syndrome was identified in a 3-generation family in which 4 of 6 moderately retarded males also had episodes of manic-depressive psychosis. ... Meloni et al. (2000) studied the same family and compared the phenotypic findings with those of Rett syndrome. Similarities included absence of language, ataxic gait, seizures, grinding of teeth, and sialorrhea. Moreover, spastic paraparesis is a frequent end-stage finding in Rett syndrome. Salient differences included absence of growth retardation, of loss of acquired purposeful hand skills, and of acquired microcephaly. Microcephaly is one of the major diagnostic criteria of Rett syndrome, in contrast with the macrocephaly in the family studied by Meloni et al. (2000).