-
Severe Dermatitis-Multiple Allergies-Metabolic Wasting Syndrome
Orphanet
Severe dermatitis-multiple allergies-metabolic wasting syndrome is a rare, genetic, epidermal disorder characterized by congenital erythroderma with severe psoriasiform dermatitis, ichthyosis, severe palmoplantar keratoderma, yellow keratosis on the hands and feet, elevated immunoglobulin E, multiple food allergies, and metabolic wasting.
-
Kagami-Ogata Syndrome
Orphanet
Kagami-Ogata syndrome is a rare genetic disease characterized by polyhydramnios (mostly due to placentomegaly), fetal macrosomia, abdominal wall defects, skeletal abnormalities (including bell-shaped thorax, coat-hanger appearance of the ribs and decreased mid to wide thorax diameter ratio in infancy), feeding difficulties and impaired swallowing, dysmorphic features (hairy forehead, full cheeks, protruding philtrum, micrognathia), developmental delay and intellectual disability.
-
Cardiocranial Syndrome, Pfeiffer Type
Orphanet
A rare, multiple congenital anomalies syndrome with intellectual disability commonly characterized by facial dysmorphism (e.g. sagittal craniosynostosis, hypertelorism, strabismus, low-set dysplastic ears, retrognathia or micrognathia, mandibular ankyloses, cleft palate, aplasia uvulae), congenital heart defects (e.g. atrioventricular septal defect, anomalous venous return), genital anomalies (e.g. cryptorchidism, microphallus), as well as growth delay and intellectual disability.
-
14q22q23 Microdeletion Syndrome
Orphanet
14q22q23 microdeletion syndrome is a rare partial deletion of the long arm of chromosome 14 characterized by ocular anomalies (anopthalmia/microphthalmia, ptosis, hypertelorism, exophthalmos), pituitary anomalies (pituitary hypoplasia/aplasia with growth hormone deficiency and growth retardation) and hand/foot anomalies (polydactyly, short digits, pes cavus).
-
Progressive Essential Tremor-Speech Impairment-Facial Dysmorphism-Intellectual Disability-Abnormal Behavior Syndrome
Orphanet
A rare genetic syndromic intellectual disability characterized by global developmental delay, moderate to severe intellectual disability, motor and language impairment, behavioral abnormalities (with mood instability, aggression, and self-mutilation), and progressive hand tremor.
-
Rhizomelic Syndrome, Urbach Type
Orphanet
Rhizomelic syndrome, Urbach type is a rare primary bone dysplasia characterized by upper limbs rhizomelia and other skeletal anomalies (e.g. short stature, dislocated hips, digitalization of the thumb with bifid distal phalanx), craniofacial features (e.g. microcephaly, large anterior fontanelle, fine and sparse scalp hair, depressed nasal bridge, high arched palate, micrognathia, short neck), congenital heart defects (e.g. pulmonary stenosis), delayed psychomotor development and mild flexion contractures of elbows.
-
Micrognathia-Recurrent Infections-Behavioral Abnormalities-Mild Intellectual Disability Syndrome
Orphanet
A rare multiple congenital anomalies/dysmorphic syndrome with intellectual disability characterized by mild global developmental delay, intellectual disability or learning difficulties, behavioral problems (like autistic, hyperactive, or aggressive behavior), variable dysmorphic craniofacial features, and abnormalities of the fingers (brachydactyly, tapering fingers, prominent interphalangeal joints).
- Severe Growth Deficiency-Strabismus-Extensive Dermal Melanocytosis-Intellectual Disability Syndrome Orphanet
-
Xq25 Microduplication Syndrome
Orphanet
A rare, X-linked, multiple congenital anomalies/dysmorphic malformation-intellectual disability syndrome characterized by developmental delay, mild to moderate intellectual disability, speech disturbance, behavioral problems (such as anxiety, hyperactivity, and aggressiveness) and mild facial dysmorphism (including facial hypotonia, thin arched eyebrows, ectropion, epicanthus, malar flatness, thick vermillion of the lips and prognathia).
-
Stag1-Related Intellectual Disability-Facial Dysmorphism-Gastroesophageal Reflux Syndrome
Orphanet
A rare genetic multiple congenital anomalies/dysmorphic syndrome characterized by global developmental delay, variable degrees of intellectual disability, and facial dysmorphism (including high nasal bridge, deep-set eyes, and wide mouth), often associated with feeding difficulties and/or gastroesophageal reflux.
-
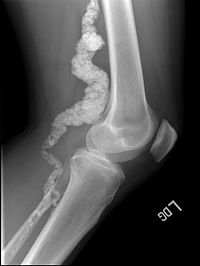
Hereditary Arterial And Articular Multiple Calcification Syndrome
Orphanet
Hereditary arterial and articular multiple calcification syndrome is a very rare genetic vascular disease of autosomal recessive inheritance, described in less than 20 patients to date, characterized by adult-onset (as early as the second decade of life) isolated calcification of the arteries of the lower extremities (including the iliac, femoral, and tibial arteries) as well as the capsule joints of the fingers, wrists, ankles and feet, and that usually manifests with mild paresthesias of the lower extremities, intense joint pain and swelling, and early onset arthritis of affected joints.
-
Leukoencephalopathy-Dystonia-Motor Neuropathy Syndrome
Orphanet
Leukoencephalopathy-dystonia-motor neuropathy syndrome is a peroxisomal neurodegenerative disorder characterized by spasmodic torticollis, dystonic head tremor, intention tremor, nystagmus, hyposmia, and hypergonadotrophic hypogonadism with azoospermia.
-
Congenital Neutropenia-Myelofibrosis-Nephromegaly Syndrome
Orphanet
Congenital neutropenia-myelofibrosis-nephromegaly syndrome is rare, genetic, primary immunodeficiency disorder characterized by severe congenital neutropenia, bone marrow fibrosis and neutrophil dysfunction which is refractory to granulocyte colony-stimulating factor, manifesting with life-threatening infections and/or deep-seated abscesses, hepato-/splenomegaly, thrombocytopenia, hypergammaglobulinemia, anemia with reticulocytosis and nephromegaly.
-
Congenital Cataract-Hearing Loss-Severe Developmental Delay Syndrome
Orphanet
Congenital cataract-hearing loss-severe developmental delay syndrome is a rare, genetic, lethal, neurometabolic disease characterized by congenital cataracts, sensorineural hearing loss, severe psychomotor developmental delay, severe, generalized muscular hypotonia, and central nervous system abnormalities (incl. cerebellar and cerebral hypoplasia, hypomyelination, wide subarachnoid spaces), in the presence of low serum copper and ceruloplasmin.
- Short Rib-Polydactyly Syndrome, Verma-Naumoff Type Orphanet
-
Ondine Syndrome
Orphanet
Congenital central hypoventilation syndrome (CCHS) is a rare disease due to a severely impaired central autonomic control of breathing and dysfunction of the autonomous nervous system.
-
Adult-Onset Multiple Mitochondrial Dna Deletion Syndrome Due To Dguok Deficiency
Orphanet
An extremely rare multiple mitochondrial DNA deletion syndrome with markedly decreased deoxyguanosine kinase (DGUOK) activity in skeletal muscle characterized by a highly variable phenotype.
-
Growth And Developmental Delay-Hypotonia-Vision Impairment-Lactic Acidosis Syndrome
Orphanet
Growth and developmental delay-hypotonia-vision impairment-lactic acidosis syndrome is a rare, genetic, mitochondrial oxidative phosphorylation disorder characterized by intrauterine growth retardation, microcephaly, hypotonia, vision impairment, speech and language delay and lactic acidosis with reduced respiratory chain activity (typically complex I).
-
Lipodystrophy
Wikipedia
The medicine is used in: adults and children above the age of two years with generalised lipodystrophy ( Berardinelli-Seip syndrome and Lawrence syndrome ) and in adults and children above the age of 12 years with partial lipodystrophy (including Barraquer-Simons syndrome ), when standard treatments have failed. [15] Volanesorsen is an Apo-CIII inhibitor [16] [17] that is currently being investigated as a potential therapeutic to reduce hypertriglycerides in Familial Partial Lipodystrophy patients in the BROADEN study. [18] Society and culture [ edit ] Lipodystrophy United is an American organization founded and run by lipodystrophy patients to support each other and raise awareness about lipodystrophy syndromes. [19] Lipodystrophy UK is a dedicated UK charity set up to support people affected by Lipodystrophy. [20] March 31 st is observed as the World Lipodystrophy Day. [21] [22] See also [ edit ] Keppen–Lubinsky syndrome Lipoedema References [ edit ] ^ Phan J, Reue K (January 2005). ... "The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline" . ... Journal of Acquired Immune Deficiency Syndromes . 33 (1): 29–33. doi : 10.1097/00126334-200305010-00005 . ... "Type 1 familial partial lipodystrophy: understanding the Köbberling syndrome". Endocrine . 54 (2): 411–421. doi : 10.1007/s12020-016-1002-x . ... "Targeting APOC3 in the familial chylomicronemia syndrome". The New England Journal of Medicine . 371 (23): 2200–6. doi : 10.1056/NEJMoa1400284 .BSCL2, AGPAT2, ZMPSTE24, POLD1, PPARG, CAVIN1, PARP2, LPIN1, EBF1, LMNA, FBN1, INSR, PIK3R1, PLIN1, CAV1, PSMB8, AKT2, EMD, IGF1R, PDGFRB, PIK3CA, WRN, CIDEC, IFIH1, LMNB2, BANF1, VCL, CSRP3, TTN, TPM1, TNNT2, TNNI3, TCAP, SDHA, TNNC1, TMPO, TAZ, SGCD, USP8, SCN5A, RAF1, ACTC1, PSMB4, PSEN2, PSEN1, PMM2, PLN, TAF1A, NEBL, ADIPOQ, PPCS, SLC29A3, GATAD1, PRDM16, CDH23, TMEM43, RNASEH2B, CLMP, GMNN, SPRTN, RNASEH2C, MYPN, OTULIN, NEXN, RBM20, RBM28, SLC25A24, BAG3, LDB3, ABCC9, CAP2, MYH6, RNASEH2A, TXNRD2, POLR3A, TREX1, ANKRD1, B4GALT7, DOLK, SYNE2, SYNE1, ATP6V0A2, SAMHD1, MYH7, ACTB, MYBPC3, DMD, KRAS, FOS, FLNA, FHL2, FHL1, FGFR1, FKTN, DSG2, DES, LEP, DDOST, CRYAB, COL3A1, ATP6V1E1, ATP6V1A, ALB, ADAR, ACTN2, LAMA4, FUCA1, PSMB9, LIPE, TNF, APOC3, FGF21, IL18, TYMS, GH1, DAPK3, ESR2, MFN2, CRP, SREBF1, RETN, CNBP, CD68, IFNG, IGF1, IL1A, HLA-B, APOE, CCL2, CXCL8, SERPINE1, ZGPAT, KCNJ6, FAS, REEP1, PCK1, ANGPTL3, AGL, CTNNB1, FETUB, RNU1-1, NPY, FITM2, NDUFA13, PTPN22, GADD45A, SMUG1, PCYT1A, MTHFR, CNOT3, DIO2, NR3C2, AGRP, NUCB2, ADIPOR1, METTL9, CACNA1S, CASP3, XYLT2, SLC28A3, ADIPOR2, CD38, CD59, PAEP, PIK3CB, CDKN1C, PNPLA2, STAB2, REG3A, CRABP2, ARL15, LMF1, ANGPTL4, MMRN1, PPARA, PDAP1, SCD, HFE, HLA-DPB1, SLC29A2, IL1B, TCP1, IL4, SLC16A2, IL6, IL7R, CCL3, SAT1, NR0B2, RP9, REN, RBP2, RARRES2, IL10, PTPRC, PTPN6, INS, LHB, LEPR, NR3C1, GRN, EGF, FSHB, SLC29A1, ESR1, FERMT2, FABP4, PIK3CD, PREP, PIK3CG, MDM2, LPL, PML, FGF19, GOT2, LPIN2, FSHR, NEDD8, LONP1, DLGAP1, SLC28A1, SLC28A2, ARTN, GPC3, HDAC3, AGPAT1
-
Benign Fasciculation Syndrome
Wikipedia
Benign fasciculation syndrome Other names Fasciculation Not Otherwise Specified Animated image of BFS in the upper eyelid of a 19-year-old male Specialty Neurology , psychiatry Benign fasciculation syndrome ( BFS ) is characterized by fasciculation (twitching) of voluntary muscles in the body. [1] The twitching can occur in any voluntary muscle group but is most common in the eyelids , arms, hands, fingers, legs, and feet. ... Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 3.1 Classification 3.2 Differential 4 Treatment 5 Prognosis 6 Research 7 References Signs and symptoms [ edit ] The main symptom of benign fasciculation syndrome is focal or widespread involuntary muscle activity ( fasciculation ). [1] The benign twitches usually have a constant location. [2] Other common symptoms are generalized fatigue or weakness, paraesthesia or numbness, and muscle cramping or spasms. [1] Anxiety and somatic symptom disorders and symptoms are commonly reported. [1] Muscle stiffness may also be present; if muscle weakness is not also present, and cramps are more severe, the stiffness may be categorized instead as cramp fasciculation syndrome . [3] Cramp fasciculation is a variant of BFS which presents with muscle pain and exercise intolerance . [2] [4] BFS symptoms are typically not accompanied by severe muscle weakness, and are typically present when the muscle is at rest. ... PMID 31174866 . ^ a b c d e f g h i j k l m Walter TR (March 2015). "Benign fasciculation syndrome". J Pain Palliat Care Pharmacother . 29 (1): 54–5. doi : 10.3109/15360288.2014.997856 . ... "Transitory stapedial myoclonus in a patient with benign fasciculation syndrome". J Laryngol Otol (Review). 127 (6): 605–6. doi : 10.1017/S0022215113000297 . ... PMID 9227959 . ^ Tzatha E, Langsdorf J, Carey B, Chin RL (March 18, 2013). "Benign fasciculation syndrome as a manifestation of small fiber neuropathy (P01.139)" .


