A number sign (#) is used with this entry because of evidence that autosomal recessive Robinow syndrome-2 (RRS2) is caused by homozygous or compound heterozygous mutation in the NXN gene (612895) on chromosome 17p13. Description Autosomal recessive Robinow syndrome-2 is a skeletal dysplasia characterized by postnatal mesomelic short stature and relative macrocephaly as well as dysmorphic facial features, including frontal bossing, hypertelorism, prominent eyes, wide short nose with anteverted nares, and triangular mouth. ... For a discussion of genetic heterogeneity of autosomal recessive Robinow syndrome, see RRS1 (268310). Clinical Features White et al. (2018) studied 2 sisters (BAB9844 and BAB9847, from family HOU3634), ages 3 years and 6 months, respectively, and an unrelated 5-year-old Turkish girl (BAB8841, from family HOU3189), who exhibited features of autosomal recessive Robinow syndrome. ... Molecular Genetics In a 5-year-old Turkish girl with autosomal recessive Robinow syndrome, who was negative for mutation in the ROR2 gene (602337), White et al. (2018) performed exome sequencing and identified homozygosity for a nonsense mutation in the NXN gene (R209X; 612895.0001) that segregated with disease in the family.
15q24 microdeletion syndrome is a rare chromosomal anomaly characterized cytogenetically by a 1.7-6.1 Mb deletion in chromosome 15q24 and clinically by pre- and post-natal growth retardation, intellectual disability, distinct facial features, and genital, skeletal, and digital anomalies. Epidemiology The prevalence of 15q24 deletion syndrome is unknown. To date, 19 cases with clinical data and detailed mapping of genomic breakpoints have been reported. ... Other congenital malformations, while rare, can be severe and include cardiovascular malformations, congenital diaphragmatic hernia, intestinal atresia, imperforate anus, and myelomeningocele (see these terms). Etiology The syndrome is caused by a microdeletion of 1.7 to 6.1 Mb in size in chromosome 15q24 which usually results from nonallelic homologous recombination (NAHR). ... Differential diagnosis Differential diagnoses include other genetic syndromes, particularly monosomy 22q11, Prader-Willi, and Noonan syndromes (see these terms).
A number sign (#) is used with this entry because of evidence that Witteveen-Kolk syndrome (WITKOS) is caused by heterozygous mutation in the SIN3A gene (607776) on chromosome 15q24. Some patients with a similar disorder have a contiguous gene deletion syndrome (chr15:72.15-73.85 Mb, NCBI36) that includes the SIN3A gene. ... Although the duplication was inherited from the healthy father, it was considered clinically significant, since the phenotype in the proband resembled the reciprocal deletion syndrome. El-Hattab et al. (2009) reported a 15-year-old boy with short stature, mild mental retardation, hypertonia, attention-deficit hyperactivity disorder, and Asperger syndrome who had a 2.6-Mb microduplication of chromosome 15q24, including the 1.75-Mb critical region. ... El-Hattab et al. (2009) identified 2 new LCR clusters involved in 15q24 deletion syndrome in addition to the 3 reported by Sharp et al. (2007) and designated them as LCR15q24A and LCR15q24C. ... The phenotype was similar to that observed in patients with chromosome 15q24 deletion syndrome, suggesting that haploinsufficiency for SIN3A is the main cause of the phenotype of that disorder.
Desbuquois syndrome (DBQD) is an osteochondrodysplasia characterized by severe micromelic dwarfism, facial dysmorphism, joint laxity with multiple dislocations, vertebral and metaphyseal abnormalities and advanced carpotarsal ossification. ... Clinical description DBQD is characterized by severe micromelic dwarfism, facial dysmorphism (round flat face, prominent eyes, midface hypoplasia, short nose, microstomia, long upper lip with flat philtrum, microretrognathia, often resulting in isolated Pierre Robin syndrome (see this term)), thoracic hypoplasia, kyphoscoliosis, severe joint laxity with dislocation, and osteopenia. ... Differential diagnosis Differential diagnosis includes autosomal dominant or recessive Larsen syndrome, Reunion island's Larsen syndrome, Catel-Manzke syndrome, chondrodysplasia with joint dislocations, gPAPP type, CHST3-related skeletal dysplasia, spondyloepiphyseal dysplasia, Omani type, diastrophic dwarfism and humerospinal dysostosis (see these terms).
The differential diagnosis of Desbuquois syndrome includes some conditions that have been lumped under the heading of Larsen syndrome (245600). Le Merrer et al. (1991) suggested that cases II and III and possibly case IV of Silverman (1972) had Desbuquois syndrome rather than Larsen syndrome. The findings in the hand in Desbuquois syndrome are particularly distinctive. ... Le Merrer et al. (1991) reported 3 patients with Desbuquois syndrome who showed deviation of digits and supernumerary metacarpal bones. ... At least one had lethal severe respiratory distress associated with small thorax and others had recurrent moderate to severe respiratory distress syndrome (RDS) during infancy. All patients had generalized joint laxity with dislocatable knees and/or femora. ... Faivre et al. (2004) provided follow-up on 4 patients (3 males and 1 female) with Desbuquois syndrome, aged 16 to 22 years. The patients had previously been described by Piussan et al. (1975) and Le Merrer et al. (1991).
Baratela et al. (2012) described 7 male patients from 6 families of different ethnic backgrounds with a syndrome of skeletal dysplasia, characteristic facial features, and developmental delay. ... In 12 patients from 10 unrelated families exhibiting a skeletal syndrome with features overlapping those of DBQD2, including 6 patients from 5 families originally reported by Baratela et al. (2012), LaCroix et al. (2019) identified homozygous and compound heterozygous mutations in the XYLT1 gene (see, e.g., 608124.0008-608124.0012).
Whilst wooly hair may occur as an isolated finding, it is important to exclude manifestations that occur in syndromic forms such as dilated cardiomyopathy and palmoplantar keratoderma (Carvajal syndrome), arrhythmogenic right ventricular cardiomyopathy and palmoplantar keratoderma (Naxos disease), or with growth failure and neurological symptoms (Menkes disease). ... Differential diagnosis Differential diagnosis includes acquired progressive curling of the hair, allotrichia circumscripta symmetrica, acquired partial kinky hair and drug-induced kinky hair. Syndromes with woolly hair should also be excluded, such as Naxos disease, Carvajal syndrome, Woolly hair-hypotrichosis-everted lower lip-outstanding ears syndrome, woolly-hair-palmoplantar keratoderma syndrome, and skin fragility-woolly hair-palmoplantar keratoderma syndrome. ... Harsh physical and chemical cosmetic treatments should be avoided. If the presence of a syndrome is suspected, an extensive internal investigation, with a detailed cardiological diagnostic examination, is necessary.
Find sources: "Adult-onset immunodeficiency syndrome" – news · newspapers · books · scholar · JSTOR ( August 2012 ) Adult-onset immunodeficiency syndrome Other names Adult-onset immunodeficiency with anti-interferon-gamma autoantibodies Adult-onset immunodeficiency syndrome is a provisional name for an immunodeficiency illness. The name is proposed in the first public study to identify the syndrome. [1] It appears to be chronic and non- contagious , affecting mainly people of Asian descent aged around 50. [2] [3] Cases first started appearing in 2004, primarily in Thailand and Taiwan . ... Retrieved 26 August 2012 . ^ Kent Sepkowitz (2012-08-22). "New Thai-Taiwanese Syndrome Is Not AIDS 2.0" . The Daily Beast .
A rare acquired immunodeficiency disorder characterized by the appearance of susceptibility to disseminated opportunistic infections (in particular, disseminated nontuberculous mycobacterial infection, salmonellosis, penicillosis, and varicella zoster virus infection) in previously healthy (HIV-negative) adults, associated with the presence of acquired autoantibodies to interferon gamma. Typical clinical manifestation includes lymphadenopathy (cervical or generalized), fever, weight loss and/or reactive skin lesions.
Adult-onset immunodeficiency with anti-interferon-gamma autoantibodies is an immunodeficiency disorder . It is associated with susceptibility to disseminated infections (dispersed throughout the body) caused by organisms that typically affect only people with weak immune systems (opportunistic pathogens). People with this disorder produce higher amounts of anti-interferon-gamma autoantibodies. These are specific immune system proteins that mistakenly target a person's own tissues. It is predominantly reported in Southeast Asians who were previously healthy.
A rare primary bone dysplasia characterized by Perthes-like pelvic anomalies (premature closure of the capital femoral epiphyses and widened femoral necks with flattened femoral heads), arthralgias of hips and knees, and occurrence of enchondromata and ecchondromata. There have been no further descriptions in the literature since 1971.
In 5 sibships and 3 generations of a family originating from Upington district of the Cape Province, South Africa, Schweitzer et al. (1971) described a new 'dyschondroplasia' in which father-son transmission was noted. The features were Perthes-like hip changes, enchondromata and ecchondromata. Some similarities to Ollier disease and Maffucci disease were noted. The latter two conditions are, however, not mendelian. INHERITANCE - Autosomal dominant SKELETAL - Enchondromata (cartilaginous tumor growing from interior of bone) - Ecchondromata (cartilaginous tumor projecting under periosteum) - Arthralgias (hips, knees) Pelvis - Premature closure of the capital femoral epiphyses - Widened femoral necks - Flattened femoral heads MISCELLANEOUS - Onset at age 5 years - Majority of cases have bilateral involvement ▲ Close
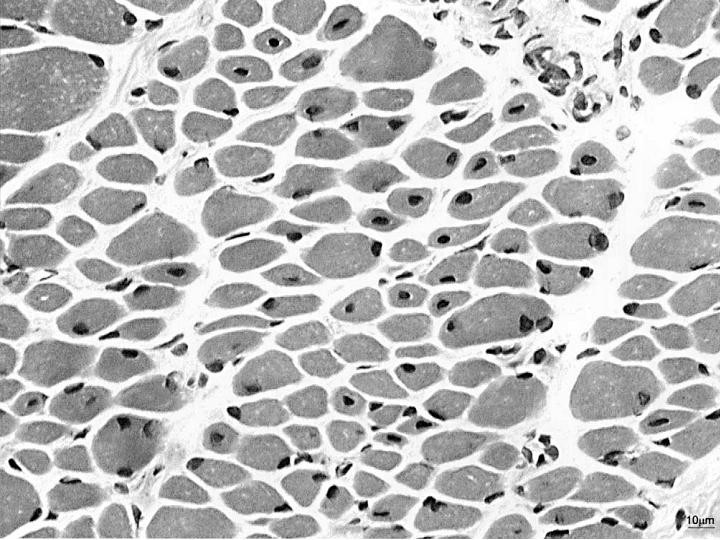
A rare group of inherited neuromuscular disorders characterized by clinical features of a congenital myopathy and centrally placed nuclei on muscle biopsy. The clinical picture and other histologic features varies according to gene involved and mode of inheritance. Epidemiology The exact prevalence and incidence of this group of diseases are unknown. For the X-linked form (XLMTM1) the incidence is estimated at 1/50,000 male birth. Clinical description These congenital myopathies are characterized by generalized muscle weakness that can range from mild to severe.
Centronuclear myopathy refers to a group of rare, inherited conditions that affect the muscles. There are three main forms of the condition that are differentiated by their pattern of inheritance: X-linked Myotubular Myopathy Autosomal Dominant Centronuclear Myopathy Autosomal Recessive Centronuclear Myopathy The cause of the condition and the associated signs and symptoms vary by subtype. For more information, click on the link of interest above. Treatment is based on the signs and symptoms present in each person and may include physical and/or occupational therapy and assistive devices to help with mobility, eating and/or breathing.
Other less common causes include hypercoagulable state, cancer, kidney transplantation, Behcet syndrome , antiphospholipid antibody syndrome or blunt trauma to the back or abdomen. [3] Treatment of RVT mainly focuses on preventing further blood clots in the kidneys and maintaining stable kidney function. ... Usually the diagnoses of RVT is first made when a nephrotic syndrome patient experiences a pulmonary embolism or a sudden decrease in kidney function or kidney failure. ... The increased loss of proteins in the urine caused by nephrotic syndrome results in lower osmotic pressure. ... Diagnosis [ edit ] CT showing dilatation and thrombosis of the left renal vein in a patient with nutcracker syndrome There are no laboratory tests used to diagnose RVT. ... Patients already suffering from nephrotic syndrome may not need to take anticoagulants.
Anaplastic large cell lymphoma (ALCL) is a rare type of Non-Hodgkins lymphoma. Lymphoma is a cancer of the lymph system, part of our immune system. Non-Hodgkins lymphoma involves abnormal growth of white blood cells, either T cells or B cells. Anaplastic large cell lymphoma is an aggressive cancer that usually involves the T-cells. Cancer cells in ALCL can be identified by their appearance under the microscope and by the presence of a tumor marker called CD30 or Ki-1. There are two types of ALCL, a type that affects mainly the skin (cutaneous ALCL) and a type that affects other body organs (systmic ALCL).
A rare and aggressive peripheral T-cell non-Hodgkin lymphoma, belonging to the group of CD30-positive lymphoproliferative disorders, which affects lymph nodes and extranodal sites. It is comprised of two sub-types, based on the expression of a protein called anaplastic lymphoma kinase (ALK): ALK positive and ALK negative ALCL. Epidemiology ALCL accounts for approximately 3% of adult non-Hodgkin lymphomas and 10% to 20% of childhood lymphomas. Its prevalence is unknown. The ALK positive subtype usually affects children and young adults. The ALK negative subtype is more commonly found in older patients over the age of 40.
A rare chronic immune-mediated polyneuropathy characterized by a progressive disabling neuropathy with marked gait disturbance primarily due to sensory ataxia with concurrent cranial neuropathies (internal or external ophthalmoplegia, dysphagia, dysarthria, or facial weakness) and anti-disialosyl IgM antibodies. Epidemiology The disease is rare with less than 100 cases reported in the literature. The disease predominantly affects males with a ratio of 3:1 (males:females). Clinical description The condition appears in adult or elderly age with a median age at onset of 55 years. The clinical picture comprises a chronic neuropathy with marked sensory ataxia and hyporeflexia/areflexia.
In 2 brothers, Abels and Reed (1973) described a disorder with both similarities to and differences from the Fanconi syndrome. One brother died in his mid-twenties after a prolonged course characterized by pancytopenia, recurrent infections, low IgA, chronic lung infections complicated by multiple bilateral pneumothoraces, osteomyelitis, and multiple cutaneous malignancies with lymph node metastases.
Congenital cataract-progressive muscular hypotonia-hearing loss-developmental delay syndrome is a rare, genetic, mitochondrial myopathy disorder characterized by congenital cataract, progressive muscular hypotonia that particularly affects the lower limbs, reduced deep tendon reflexes, sensorineural hearing loss, global development delay and lactic acidosis.
A number sign (#) is used with this entry because of evidence that mitochondrial progressive myopathy with congenital cataract, hearing loss, and developmental delay is caused by homozygous mutation in the GFER gene (600924) on chromosome 16p13. One such family has been reported. Clinical Features Di Fonzo et al. (2009) reported a consanguineous Moroccan family including 3 children with congenital cataract, muscular hypotonia, sensorineural hearing loss, and developmental delay. The first child presented in the first month of life with congenital cataract and axial hypotonia. He did not learn to walk until 2 years of age or speak until 3 years of age. At the age of 12 he developed progressive hearing loss and bilateral ptosis.
Fetal encasement syndrome is a rare, lethal developmental defect during embryogenesis characterized by severe fetal malformations, including craniofacial dysmorphism (abnormal cyst in the cranial region, hypoplastic eyeballs, two orifices in the nasal region separated by a nasal septum, abnormal orifice replacing the mouth), omphalocele and immotile, hypoplastic limbs encased under an abnormal, transparent, membrane-like skin.
A number sign (#) is used with this entry because of evidence that fetal encasement syndrome, an autosomal recessive condition, is caused by homozygous mutation in the CHUK (600664) gene on chromosome 10q24. ... Kalay et al. (2012) noted similarities between fetal encasement syndrome and the Bartsocas-Papas phenotype (263650) caused by mutation in the RIPK4 gene (605706).
A rare X-linked syndromic intellectual disability which in symptomatic, female carriers is characterized by a highly variable phenotype including facial dysmorphisms (prominent forehead, hypertelorism, down-slanting palpebral fissures, epicanthic folds, thick lips with everted lower vermilion, thick nasal alae, and septum), short hands with tapering fingers, short stature and skeletal findings (progressive kyphoscoliosis).
Early-onset spastic ataxia-myoclonic epilepsy-neuropathy syndrome is a rare hereditary spastic ataxia disorder characterized by childhood onset of slowly progressive lower limb spastic paraparesis and cerebellar ataxia (with dysarthria, swallowing difficulties, motor degeneration), associated with sensorimotor neuropathy (including muscle weakness and distal amyotrophy in lower extremities) and progressive myoclonic epilepsy.
A number sign (#) is used with this entry because of evidence that autosomal recessive spastic ataxia-5 (SPAX5) is caused by homozygous mutation in the AFG3L2 gene (604581) on chromosome 18p11. Heterozygous mutation in the AFG3L2 gene causes autosomal dominant spinocerebellar ataxia-28 (SCA28; 610246). Description Spastic ataxia-5 (SPAX5) is an autosomal recessive neurodegenerative disorder characterized by early-onset spasticity resulting in significantly impaired ambulation, cerebellar ataxia, oculomotor apraxia, dystonia, and myoclonic epilepsy (summary by Pierson et al., 2011). For a discussion of genetic heterogeneity of spastic ataxia, see SPAX1 (108600). Clinical Features Pierson et al. (2011) reported 2 brothers, born of consanguineous Hispanic parents from Colombia, with early-onset spinocerebellar ataxia with spasticity and myoclonic epilepsy.
Hemophagocytic syndrome (HPS) is a rare immune disease (see this term) and a potentially life-threatening disorder characterized by cytokine storm and overwhelming inflammation causing fever, hepatosplenomegaly, cytopenia, hypertriglyceridemia, hyperferritinemia, and hemophagocytosis in bone marrow, liver, spleen or lymph nodes.
It is classified as one of the cytokine storm syndromes. There are inherited and non-inherited (acquired) causes of hemophagocytic lymphohistiocytosis (HLH). ... Acquired HLH may have decreased, normal, or increased NK cell activity. [ citation needed ] Griscelli syndrome [ edit ] A major differential diagnosis of HLH is Griscelli syndrome (type 2). ... Type 2 has mutations in RAB27A and haemophagocytic syndrome, with abnormal T-cell and macrophage activation. ... "COVID-19: consider cytokine storm syndromes and immunosuppression" . The Lancet . 395 (10229): 1033–1034. doi : 10.1016/S0140-6736(20)30628-0 . ... "Autoimmune lymphoproliferative syndrome misdiagnosed as hemophagocytic lymphohistiocytosis" .