Corneal opacities, not impairing visual acuity, were seen in 14 of 28 males by slit-lamp examination. Garcia Perez and Crespo (1981) and Stoll et al. (1983) reported 2 families containing 5 boys with the combination of hypertrophic pyloric stenosis and X-linked recessive ichthyosis.
Clinical Features A common form of X-linked ichthyosis (308100), also known as steroid sulfatase deficiency, is caused by mutation in the STS gene (300747). Robledo et al. (1995) described a Sardinian kindred in which congenital ichthyosis was associated with normal levels of steroid sulfatase and a normal pattern on Southern blot analysis suggesting the presence of an intact STS gene. Mapping Although the pedigree pattern in the kindred with ichthyosis studied by Robledo et al. (1995) was entirely consistent with X-linked recessive inheritance, the disorder was found to segregate independently of genetic polymorphisms detected by probes mapping to Xp22.3, where the STS gene maps. The search for linkage to markers elsewhere on the X chromosome had not been successful. Robledo et al. (1995) concluded that there may be a form of X-linked ichthyosis due to some mechanism other than STS deficiency.
X-linked ichthyosis is a disorder in which the skin cells are produced at a normal rate but they do not separate properly at the surface of the stratum corneum (the outermost layer of the skin). This slows the rate of shedding of the skin cells, resulting in a build-up of scales. The scales of X-linked ichthyosis are often dark and usually cover only a portion of the body. Typically, the trunk and back of the neck are more likely to be affected. Scales are usually not found on the face, scalp, palms of the hands, and soles of the feet.
There was no difference in the frequency of the gln27-to-glu (Q27E; 109690.0002) and thr164-to-ile (T164I; 109690.0003) polymorphisms between the 2 groups. In a metaanalysis of 28 published studies, Contopoulos-Ioannidis et al. (2005) confirmed the association between the gly16 polymorphism and nocturnal asthma, but found no association between the R16G or Q27E variants and asthma susceptibility overall or bronchial hyperresponsiveness.
Using an antibody array to assess the level of 28 receptors in the livers of 2 substrains of BALB/c mice, Kaushansky et al. (2015) determined that 9 receptors, including EphA2, were present at elevated levels in the substrain that is more susceptible to the murine malaria parasite, Plasmodium yoelii.
A life-threatening parasitic disease caused by Plasmodium ( P. ) parasites that are transmitted by Anophles mosquito bites to humans and is typically clinically characterized by attacks of fever, headache, chills and vomiting.
Malaria is a serious and sometimes fatal disease caused by a parasite that commonly infects a certain type of mosquito which feeds on humans. Infection with malaria parasites may result in a wide variety of symptoms, ranging from absent or very mild symptoms to severe disease and even death. People who get malaria are typically very sick with high fevers, shaking chills, and flu-like illness. In general, malaria is a curable disease if diagnosed and treated promptly and correctly. Treatment depends on many factors including disease severity, the species of malaria parasite causing the infection and the part of the world in which the infection was acquired.
The sporozoites are injected into the skin, in the saliva, when the mosquito takes a subsequent blood meal. [28] Only female mosquitoes feed on blood; male mosquitoes feed on plant nectar and do not transmit the disease. ... However, more research need to establish its efficacy as a reliable treatment. [117] Artesunate plus mefloquine performs better than mefloquine alone in treating uncomplicated falciparum malaria in low transmission settings. [118] There is limited data to show atovaquone-proguanil is more effective than chloroquine, amodiaquine, and mefloquine in treating falciparum malaria. [119] Azithromycin monotherapy or combination therapy has not shown effectiveness in treating plasmodium or vivax malaria. [120] Amodiaquine plus sulfadoxine-pyrimethamine may achieve less treatment failures when compared to sulfadoxine-pyrimethamine alone in uncomplicated falciparum malaria. [121] There is insufficient data on chlorproguanil-dapsone in treating uncomplicated falciparum malaria. [122] The addition of primaquine with artemisinin-based combination therapy for falciparum malaria reduces its transmission at day 3-4 and day 8 of infection. [123] Sulfadoxine-pyrimethamine plus artesunate is better than sulfadoxine-pyrimethamine plus amodiaquine in controlling treatment failure at day 28. However, the latter is better than the former in reducing gametocytes in blood at day 7. [124] Infection with P. vivax , P. ovale or P. malariae usually does not require hospitalisation.
Overview Malaria is a disease caused by a parasite. The parasite is spread to humans through the bites of infected mosquitoes. People who have malaria usually feel very sick with a high fever and shaking chills. While the disease is uncommon in temperate climates, malaria is still common in tropical and subtropical countries. Each year nearly 290 million people are infected with malaria, and more than 400,000 people die of the disease. To reduce malaria infections, world health programs distribute preventive drugs and insecticide-treated bed nets to protect people from mosquito bites.
Targeted analysis for the following pathogenic variants (see Table 8) may be considered. c.562_564delTTC (p.Phe188del): French-Canadian founder variant Accounts for 28% of individuals with HHH syndrome [Debray et al 2008, Martinelli et al 2015] c.535C>T (p.Arg179Ter): Japanese and Middle Eastern founder variant Accounts for 16% of individuals with HHH syndrome [Debray et al 2008, Martinelli et al 2015] A hyperammonemia or urea cycle disorder multigene panel that includes SLC25A15 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype.
Ornithine translocase deficiency is an inherited disorder that causes ammonia and other substances to build up (accumulate) in the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia. Ornithine translocase deficiency varies widely in its severity and age of onset. Affected infants show signs and symptoms of ornithine translocase deficiency within days after birth.
This article needs additional citations for verification . Please help improve this article by adding citations to reliable sources . Unsourced material may be challenged and removed. Find sources: "Ornithine translocase deficiency" – news · newspapers · books · scholar · JSTOR ( March 2008 ) ( Learn how and when to remove this template message ) Ornithine translocase deficiency Other names HHH syndrome, ORNT1 deficiency, ornithine carrier deficiency, triple H syndrome Ornithine Ornithine translocase deficiency , also called hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome , [1] is a rare autosomal recessive [2] urea cycle disorder affecting the enzyme ornithine translocase , which causes ammonia to accumulate in the blood, a condition called hyperammonemia . Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia. Contents 1 Pathophysiology 2 Diagnosis 3 Treatment 4 See also 5 References 6 Further reading 7 External links Pathophysiology [ edit ] Ornithine translocase deficiency has an autosomal recessive pattern of inheritance.
A rare, genetic disorder of urea cycle metabolism characterized by either a neonatal-onset with manifestations of lethargy, poor feeding, vomiting and tachypnea or, more commonly, presentations in infancy, childhood or adulthood with chronic neurocognitive deficits, acute encephalopathy and/or coagulation defects or other chronic liver dysfunction. Epidemiology More than 100 cases have been reported in the literature to date. The prevalence in Northern Saskatchewan, Canada is especially high due to a founder effect and is estimated in this population at 1/1550 live births. Clinical description Age of onset can range from the neonatal period to adulthood and a wide phenotypic spectrum is noted. The neonatal presentation usually begins a few days after birth with lethargy, somnolence, refusal to feed, vomiting, tachypnea with respiratory alkalosis, and/or seizures.
A number sign (#) is used with this entry because of evidence that the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome is caused by homozygous mutation in the SLC25A15 gene (603861), which encodes the mitochondrial ornithine transporter, on chromosome 13q14. Clinical Features Shih et al. (1969) reported a child with mental retardation and myoclonic seizures associated with hyperornithinemia, hyperammonemia, and homocitrullinemia. The findings were consistent with an inherited disorder of amino acid metabolism. Rodes et al. (1987) reported a family in which 2 brothers and a sister were affected with HHH syndrome. One patient had progressive spastic paraparesis. At least 2 of the individuals voluntarily avoided a high protein diet.
Molecular Genetics Grompe et al. (1994) found that 100% of patients with tyrosinemia type I from the Saguenay-Lac-Saint-Jean region of Quebec and 28% of TYRSN1 patients from other regions of the world carry a splice donor site mutation in intron 12 of the FAH gene (613871.0003).
Tyrosinemia type 1 (HTI) is an inborn error of tyrosine catabolism caused by defective activity of fumarylacetoacetate hydrolase (FAH) and is characterized by progressive liver disease, renal tubular dysfunction, porphyria-like crises and a dramatic improvement in prognosis following treatment with nitisinone. Epidemiology Birth incidence is 1/100,000 in most areas but is more common is some regions, notably in Québec, Canada. Clinical description HT1 is clinically heterogenous. Symptoms may start during the first few months (acute type), in second half of the first year (subacute type) or in the following years up to adulthood (chronic type). In the acute type, manifestations of hepatic failure predominate (bleeding diathesis, hypoglycemia, ascites etc) with frequent sepsis and rapid deterioration. Mild proximal tubular disease is usually present. Subacute type manifests a similar but less severe clinical picture presenting usually with hepatomegaly or hypophosphatemic rickets (due to tubular dysfunction).
Tyrosinemia type 1 is a genetic disorder characterized by elevated blood levels of the amino acid tyrosine, a building block of most proteins. This condition is caused by a shortage of the enzyme fumarylacetoacetate hydrolase, one of the enzymes required for the multi-step process that breaks down tyrosine. This enzyme shortage is caused by mutations in the FAH gene. Symptoms usually appear in the first few months of life and include failure to thrive , diarrhea, vomiting, jaundice, cabbage-like odor, and increased tendency to bleed (particularly nosebleeds). Tyrosinemia type I can lead to liver and kidney failure, softening and weakening of the bones, problems affecting the nervous system, and an increased risk of liver cancer. This condition is inherited in an autosomal recessive manner. Treatment should be started as soon as the condition is diagnosed and includes a diet restricted in tyrosine and phenylalanine along with nitisinone, a medication that blocks the second step in the tyrosine degradation pathway.
Summary Clinical characteristics. Untreated tyrosinemia type I usually presents either in young infants with severe liver involvement or later in the first year with liver dysfunction and renal tubular dysfunction associated with growth failure and rickets. Untreated children may have repeated, often unrecognized, neurologic crises lasting one to seven days that can include change in mental status, abdominal pain, peripheral neuropathy, and/or respiratory failure requiring mechanical ventilation. Death in the untreated child usually occurs before age ten years, typically from liver failure, neurologic crisis, or hepatocellular carcinoma. Combined treatment with nitisinone and a low-tyrosine diet has resulted in a greater than 90% survival rate, normal growth, improved liver function, prevention of cirrhosis, correction of renal tubular acidosis, and improvement in secondary rickets. Diagnosis/testing. Tyrosinemia type I results from deficiency of the enzyme fumarylacetoacetase (FAH).
Tyrosinemia is a genetic disorder characterized by disruptions in the multistep process that breaks down the amino acid tyrosine, a building block of most proteins. If untreated, tyrosine and its byproducts build up in tissues and organs, which can lead to serious health problems. There are three types of tyrosinemia, which are each distinguished by their symptoms and genetic cause. Tyrosinemia type I, the most severe form of this disorder, is characterized by signs and symptoms that begin in the first few months of life. Affected infants fail to gain weight and grow at the expected rate (failure to thrive) due to poor food tolerance because high-protein foods lead to diarrhea and vomiting.
Tyrosinemia type I Other names Hereditary Tyrosinemia type I, HT1 Mutation of enzyme fumarylacetoacetate hydrolase (FAH) in the tyrosine catabolic pathway Specialty Hepatology, nephrology, neurology Symptoms Failure to thrive, enlarged liver, fever, vomiting, diarrhea Usual onset Variable, usually with the first 2 years of life Duration Life long Causes Genetic (autosomal recessive) Diagnostic method Dried blood spot testing, urinalysis, genetic testing Treatment Dietary restrictions, Nitisinone, liver transplantation Medication Nitisinone Prognosis 93% survival rate at six years with treatment Frequency 1 in 1,850 (Saguenay-Lac Saint-Jean region, Quebec) Tyrosinemia type I is a genetic disorder that disrupts the metabolism of the amino acid tyrosine , resulting in damage primarily to the liver along with the kidneys and peripheral nerves . [1] The inability of cells to process tyrosine can lead to chronic liver damage ending in liver failure , as well as renal disease and rickets . Symptoms such as poor growth and enlarged liver are associated with the clinical presentation of the disease. [2] Clinical manifestation of disease occurs typically within the first two years of life. The severity of the disease is correlated with the timing of onset of symptoms, earlier being more severe. [1] Tyrosinemia type I is an autosomal recessive disorder caused by mutations in the both copies of the gene encoding the enzyme fumarylacetoacetate hydrolase (FAH) . FAH is a metabolic enzyme that catalyzes the conversion of fumarylacetoacetate to fumarate and acetoacetate . It is expressed primarily in the liver and kidney. Loss of FAH activity results in the accumulation of certain metabolic intermediates in the tyrosine catabolic pathway . [2] These compounds are toxic to cells and lead to differential gene expression and apoptosis in high concentrations. [2] HT1 is diagnosed when elevated levels of succinylacetone (SA), one of the metabolites in this pathway, is detected in blood and urine samples . [1] While there is no cure for tyrosinemia type I, management of the disease is possible utilizing dietary restrictions and medications .
In the 7-generation family studied by Murrell et al. (1997), a limited genomic screen by use of DNA samples from 28 family members localized the gene for this disorder to a 3-cM region on chromosome 17, between markers THRA1 (190120) (which maps to 17q11.2) and D17S791.
A number sign (#) is used with this entry because Pick disease, which belongs to a class of neurodegenerative disorders known as frontotemporal dementias (FTD; see 600274), can be caused by heterozygous mutation in the MAPT gene (157140) on chromosome 17q21. Some cases of Pick disease are caused by heterozygous mutation in the presenilin-1 gene (PSEN1; 104311) on chromosome 14q24. Description Pick disease refers to the neuropathologic finding of 'Pick bodies,' which are argyrophilic, intraneuronal inclusions, and 'Pick cells,' which are enlarged neurons. The clinical correlates of Pick disease of brain include those of frontotemporal dementia, which encompass the behavioral variant of FTD, semantic dementia, and progressive nonfluent aphasia (summary by Piguet et al., 2011). Kertesz (2003) suggested the term 'Pick complex' to represent the overlapping syndromes of FTD, primary progressive aphasia (PPA), corticobasal degeneration (CBD), progressive supranuclear palsy (601104), and FTD with motor neuron disease.
Semantic dementia (SD) is a form of frontotemporal dementia (FTD; see this term), characterized by the progressive, amodal and profound loss of semantic knowledge (combination of visual associative agnosia, anomia, surface dyslexia or dysgraphia and disrupted comprehension of word meaning) and behavioral abnormalities, attributable to the degeneration of the anterior temporal lobes.
Main articles: Aphasia and Primary progressive aphasia Semantic dementia Other names semantic variant primary progressive aphasia Specialty Neurology Semantic dementia ( SD ), also known as semantic variant primary progressive aphasia (svPPA), is a progressive neurodegenerative disorder characterized by loss of semantic memory in both the verbal and non-verbal domains. However, the most common presenting symptoms are in the verbal domain (with loss of word meaning). [1] [2] [3] Semantic dementia is a disorder of semantic memory that causes patients to lose the ability to match words or images to their meanings. [4] However, it is fairly rare for patients with semantic dementia to develop category specific impairments, though there have been document cases of it occurring. [5] Typically, a more generalized semantic impairment results form dimmed semantic representations in the brain. [6] SD is one of the three canonical clinical syndromes associated with frontotemporal lobar degeneration (FTLD), with the other two being frontotemporal dementia and progressive nonfluent aphasia . SD is a clinically defined syndrome but is associated with predominantly temporal lobe atrophy (left greater than right) and hence is sometimes called temporal variant FTLD (tvFTLD). [7] SD is one of the three variants of primary progressive aphasia (PPA), which results from neurodegenerative disorders such as FTLD or Alzheimer's disease . It is important to note the distinctions between Alzheimer’s disease and semantic dementia with regard to types of memory affected. In general, Alzheimer’s disease is referred to as disorder affecting mainly episodic memory, defined as the memory related to specific, personal events distinct for each individual.
Genotype/Phenotype Correlations Cailloux et al. (2000) investigated 52 PMD and 28 SPG families without large PLP duplications or deletions by PCR amplification and sequencing of the 7 coding regions and splice sites of the PLP1 gene.
Pelizaeus-Merzbacher disease (PMD) is an X-linked leukodystrophy characterized by developmental delay, nystagmus, hypotonia, spasticity, and variable intellectual deficit. It is classified into three sub-forms based on the age of onset and severity: connatal, transitional, and classic PMD (see these terms). Epidemiology The estimated prevalence is 1/400,000. PMD affects males but some female heterozygotes presenting with a milder phenotype have also been reported (PMD in female carriers; see this term). Clinical description The disease has a broad clinical spectrum. The connatal form is the most severe form presenting, since birth, with hypotonia, nystagmus, respiratory distress, and stridor, with subsequent motor and cognitive delay and spastic quadriparesis. The classic form manifests during the first 2 months of life with nystagmus and hypotonia which is progressively replaced by spasticity.
Pelizaeus-Merzbacher disease is an inherited condition involving the brain and spinal cord (central nervous system) that primarily affects males. This disease is one of a group of genetic disorders called leukodystrophies. Leukodystrophies are conditions that involve abnormalities of the nervous system's white matter, which consists of nerve fibers covered by a fatty substance called myelin . Myelin insulates nerve fibers and promotes the rapid transmission of nerve impulses. In particular, Pelizaeus-Merzbacher disease involves hypomyelination, which means that the nervous system has a reduced ability to form myelin.
Fahmy et al. (1969) observed a brother and sister (out of a sibship of 11), offspring of first-cousin parents, with a slowly progressive neurologic disorder that began in early childhood. Electron microscopic studies of sural nerve showed unique rod-shaped bodies in Schwann cells. The clinical picture was similar to that of Pelizaeus-Merzbacher disease (312080). Neuro - Cerebral sclerosis Lab - Rod-shaped bodies in sural nerve Schwann cells on electron microscopy Inheritance - Autosomal recessive ▲ Close
The null syndrome is part of the Pelizaeus-Merzbacher disease (PMD; see this term) spectrum and is characterized by mild PMD features associated with demyelinating peripheral neuropathy. Epidemiology Its prevalence is unknown. It predominantly affects males. Clinical description The disease manifests during childhood. Patients may have mild developmental delay, delayed sitting and walking beginning usually in the first 2 to 3 years of life, later associated with mild peripheral neuropathy, mild spastic quadriparesis, hyperreflexia, Babinski signs, ataxia, and/or mild intellectual deficit. Patients do not have nystagmus. Although they usually are ambulatory during childhood and have good speech, patients with the null syndrome tend to decline more rapidly beginning in adolescence or early adulthood compared to patients with other PMD forms. Etiology The syndrome is due to null mutations of the PLP1 gene (on Xq22) that cause hypomyelination of the central nervous system.
The connatal form of Pelizaeus-Merzbacher disease (PMD) is the most severe form of PMD (see this term). Epidemiology PMD has an estimated prevalence of 1/400,000. The connatal form accounts for approximately 10 to 15 % of all cases of PMD. It predominantly affects males. Clinical description Connatal PMD presents, from birth, with hypotonia, nystagmus, respiratory distress, stridor, feeding difficulties and sometimes seizures. Subsequently, there is profound motor and cognitive delay and spastic quadriparesis. Patients never learn to walk, have limited language skills and usually die from respiratory complications by the second decade.
X-linked leukodystrophy Pelizaeus–Merzbacher disease Pelizaeus–Merzbacher disease is inherited in an x-linked recessive manner [1] Specialty Neurology Pelizaeus – Merzbacher disease is an X-linked neurological disorder that damages oligodendrocytes in the central nervous system . It is caused by mutations in proteolipid protein 1 ( PLP1 ), a major myelin protein. It is characterized by hypomyelination and belongs to a group of genetic diseases referred to as leukodystrophies . [2] Contents 1 Signs and symptoms 2 Cause 3 Diagnosis 3.1 Classification 4 Treatment 5 Research 6 See also 7 References 8 Further reading 9 External links Signs and symptoms [ edit ] Hallmark signs and symptoms of Pelizaeus–Merzbacher disease include little or no movement in the arms or legs, respiratory difficulties, and characteristic horizontal movements of the eyes left to right. [ citation needed ] The onset of Pelizaeus–Merzbacher disease is usually in early infancy. The most characteristic early signs are nystagmus (rapid, involuntary, rhythmic motion of the eyes) and low muscle tone . Motor abilities are delayed or never acquired, mostly depending upon the severity of the mutation.
Pelizaeus-Merzbacher disease is a disorder that affects the brain and spinal cord. It is a type of leukodystrophy and is characterized by problems with coordination, motor skills, and learning. The age of onset and the severity of the symptoms varies greatly depending on the type of disease. It is caused by an inability to form myelin due to mutations in the PLP1 gene. It is passed through families in an X-linked recessive pattern. The condition primarily affects males.
Investigation of family relatives allowed detection of a de novo mutation in 16 of 76 2-generation and 28 of 34 3-generation families. On the basis of these data, Becker et al. (1996) estimated the male:female ratio of mutation frequencies (k) to be 3.6. ... These results confirmed predictions about the efficacy of the mRNA-based method suggested by Naylor et al. (1991), and also excluded hypotheses proposing that mutations outside the F8 gene are responsible for a large proportion of severe hemophilia A. Of the 28 patients reported by Naylor et al. (1993), 5 had mild or moderate disease and all had a missense mutation.
Severe hemophilia A is a form of hemophilia A (see this term) characterized by a large deficiency of factor VIII leading to frequent spontaneous hemorrhage and abnormal bleeding as a result of minor injuries, or following surgery or tooth extraction. Epidemiology Severe hemophilia A accounts for around 40% of all cases of hemophilia A. Clinical description The biological activity of factor VIII is below 1%. Etiology The disorder is caused by mutations in the F8 gene (Xq28) encoding coagulation factor VIII. Genetic counseling Transmission is X-linked recessive.
During fetal development, apoptosis was localized mainly to primary oocytes and was highest between weeks 14 and 28, decreasing thereafter toward term.
Chang et al. (1994) identified a somatic ser120-to-gly (S120G) change in 6 of 28 advanced neuroblastomas, but in none of 22 low-grade tumors or in control tissues.
A number sign (#) is used with this entry because susceptibility to neuroblastoma-3 (NBLST3) is conferred by germline or somatic mutations in the ALK gene (105590) on chromosome 2p23. For a general phenotypic description and a discussion of genetic heterogeneity of neuroblastoma, see NBLST1 (256700). Molecular Genetics Mosse et al. (2008) identified 3 separate germline missense mutations in the tyrosine kinase domain of the ALK gene that segregated with the disease in 8 separate families with neuroblastoma. There was incomplete penetrance. Resequencing in 194 high-risk neuroblastoma the samples showed somatically acquired mutations in the tyrosine kinase domain in 12.4% of samples. Nine of the 10 mutations mapped to critical regions of the kinase domain and were predicted with high probability to be oncogenic drivers.
For a general phenotypic description and a discussion of genetic heterogeneity of neuroblastoma, see NBLST1 (256700). Mapping In a genomewide analysis of 397 patients with high-risk aggressive neuroblastoma derived from the 1,032 patients in a study by Maris et al. (2008) and 2,043 controls, Capasso et al. (2009) found a significant association with 6 SNPs at chromosome 2q35 within the BARD1 locus (601593) (p = 2.35 x 10(-9) to 2.25 x 10(-8)). The associations were confirmed in a second series of 189 high-risk cases and 1,178 controls (p = 7.90 x 10(-7) to 2.77 x 10(-4)). Testing of the 2 most significant SNPs (rs6435862 and rs3768716) in 2 additional independent high-risk neuroblastoma case series yielded a combined allelic odds ratio of 1.68 for each SNP (p = 8.65 x 10(-18) and 2.74 x 10(-16), respectively). These data suggested that common variation in the BARD1 gene may contribute to the etiology of aggressive human neuroblastoma.
Neuroblastoma is a tumor that develops from neuroblasts (immature nerve tissue) in an infant or child, usually before the age of 5. It most often develops in infancy and may be diagnosed in the first month of life. The tumor most often develops in the adrenal gland , but may develop in the neck, chest, or spinal cord. It is considered an aggressive tumor because it often spreads to other parts of the body ( metastasizes ). In most cases, it has spread by the time it is diagnosed. A neuroblastoma can cause a variety of signs and symptoms, including a lump where the tumor is growing, bone pain, diarrhea, and various neurological symptoms.
Neuroblastoma is a malignant tumor of neural crest cells, the cells that give rise to the sympathetic nervous system, which is observed in children. Epidemiology It represents about 10% of solid tumors in infants and children under the age of 15, with an annual incidence of about 1/70,000 in children in this class of age. Clinical description In 90% of cases the neuroblastoma is diagnosed before the age of five. The clinical presentation of neuroblastoma is very variable and depends on the stage and location of the tumor, which can develop at any site in the sympathetic nervous system (around 80% of cases develop in the abdomen). Localized forms are discovered fortuitously or are revealed by the presence of an abdominal or thoracic mass that can be associated with pain.
Overview Neuroblastoma is a cancer that develops from immature nerve cells found in several areas of the body. Neuroblastoma most commonly arises in and around the adrenal glands, which have similar origins to nerve cells and sit atop the kidneys. However, neuroblastoma can also develop in other areas of the abdomen and in the chest, neck and near the spine, where groups of nerve cells exist. Neuroblastoma most commonly affects children age 5 or younger, though it may rarely occur in older children. Some forms of neuroblastoma go away on their own, while others may require multiple treatments.
Homer Wright pseudorosettes are tumor cells around the neuropil , not to be confused with a true rosettes, which are tumor cells around an empty lumen. [26] They are also distinct from the pseudorosettes of an ependymoma which consist of tumor cells with glial fibrillary acidic protein (GFAP)–positive processes tapering off toward a blood vessel (thus a combination of the two). [27] A variety of immunohistochemical stains are used by pathologists to distinguish neuroblastomas from histological mimics, such as rhabdomyosarcoma , Ewing's sarcoma , lymphoma and Wilms' tumor . [28] Neuroblastoma is one of the peripheral neuroblastic tumors (pNTs) that have similar origins and show a wide pattern of differentiation ranging from benign ganglioneuroma to stroma -rich ganglioneuroblastoma with neuroblastic cells intermixed or in nodules, to highly malignant neuroblastoma. ... Updated results of the population based controlled trial in Germany". Cancer Letters . 197 (1–2): 19–28. doi : 10.1016/S0304-3835(03)00077-6 . ... Retrieved 2008-07-30 . ^ Darshak Sanghavi, "Screen Alert: How an Ounce of RX Prevention can Cause a Pound of Hurt" Archived 2006-12-01 at the Wayback Machine , Slate magazine, November 28, 2006 ^ Johnson E, Dean SM, Sondel PM (December 2007). ... PMID 9149032 . ^ Ladenstein R, Pötschger U, Hartman O, Pearson AD, Klingebiel T, Castel V, et al. (June 2008). "28 years of high-dose therapy and SCT for neuroblastoma in Europe: lessons from more than 4000 procedures" .
For a general phenotypic description and a discussion of genetic heterogeneity of neuroblastoma, see NBLST1 (256700). Mapping Maris et al. (2008) provided evidence for 1 or more candidate neuroblastoma susceptibility genes on chromosome 6p22. Among 1,032 neuroblastoma patients and 2,043 controls of European descent, the authors observed an association between disease and 3 SNPs on chromosome 6p22: rs6939340, rs4712653, and rs9295536, yielding p values of 1.71 x 10(-9) to 7.01 x 10(-10) (allelic odds ratio of 1.39 to 1.40). The findings suggested that common genetic variants may predispose to increased risk for neuroblastic malignant transformation. In a genomewide analysis of 397 patients with high-risk aggressive neuroblastoma derived from the 1,032 patients in the study of Maris et al. (2008) and 2,043 controls, Capasso et al. (2009) confirmed the association of the 3 SNPs at 6p22 identified by Maris et al. (2008) as being more significantly associated with a high-risk subtype of neuroblastoma.
For a general phenotypic description and a discussion of genetic heterogeneity of neuroblastoma, see NBLST1 (256700). Mapping Diskin et al. (2009) described the identification of a common copy number variation (CNV) at chromosome 1q21.1 associated with neuroblastoma in a discovery set of 846 Caucasian patients with neuroblastoma and 803 controls. The findings were confirmed in 2 independent replication sets comprising 595 cases and 3,357 controls. They first observed a hemizygous approximately 300-kb deletion at 1q21.1 that occurred in 15.6% of cases but in only 9.1% of controls. The difference in hemizygous deletion frequency was significant at p = 1.83 x 10(-19).
A number sign (#) is used with this entry because susceptibility to neuroblastoma-2 (NBLST2) is conferred by germline mutations in the PHOX2B gene (603851) on chromosome 4p13. For a general phenotypic description and a discussion of genetic heterogeneity of neuroblastoma, see NBLST1 (256700). See also congenital central hypoventilation syndrome (CCHS; 209880), which is also caused by mutation in the PHOX2B gene. Patients with CCHS have a high predisposing risk of developing a tumor of the sympathetic nervous system, as indicated by a 5 to 10% occurrence of neuroblastoma, ganglioneuroblastoma, and ganglioneuroma (Rohrer et al., 2002; Amiel et al., 2003). Clinical Features Trochet et al. (2004) reported a family with neuroblastoma.
For a general phenotypic description and a discussion of genetic heterogeneity of neuroblastoma, see NBLST1 (256700). Mapping To identify genetic risk factors for neuroblastoma, Wang et al. (2011) performed a genomewide association study on 2,251 patients and 6,097 control subjects of European ancestry from 4 case series. Wang et al. (2011) reported a significant association with LMO1 (186921) at 11p15.4 (rs110419, combined p = 5.2 x 10(-16), odds ratio (OR) risk allele = 1.34, 95% CI 1.25-1.44). The signal was enriched in the subset of patients with the most aggressive form of the disease. LMO1 encodes a cysteine-rich transcriptional regulator, and its paralogs LMO2 (180385), LMO3 (180386), and LMO4 (603129) have each been implicated in cancer.
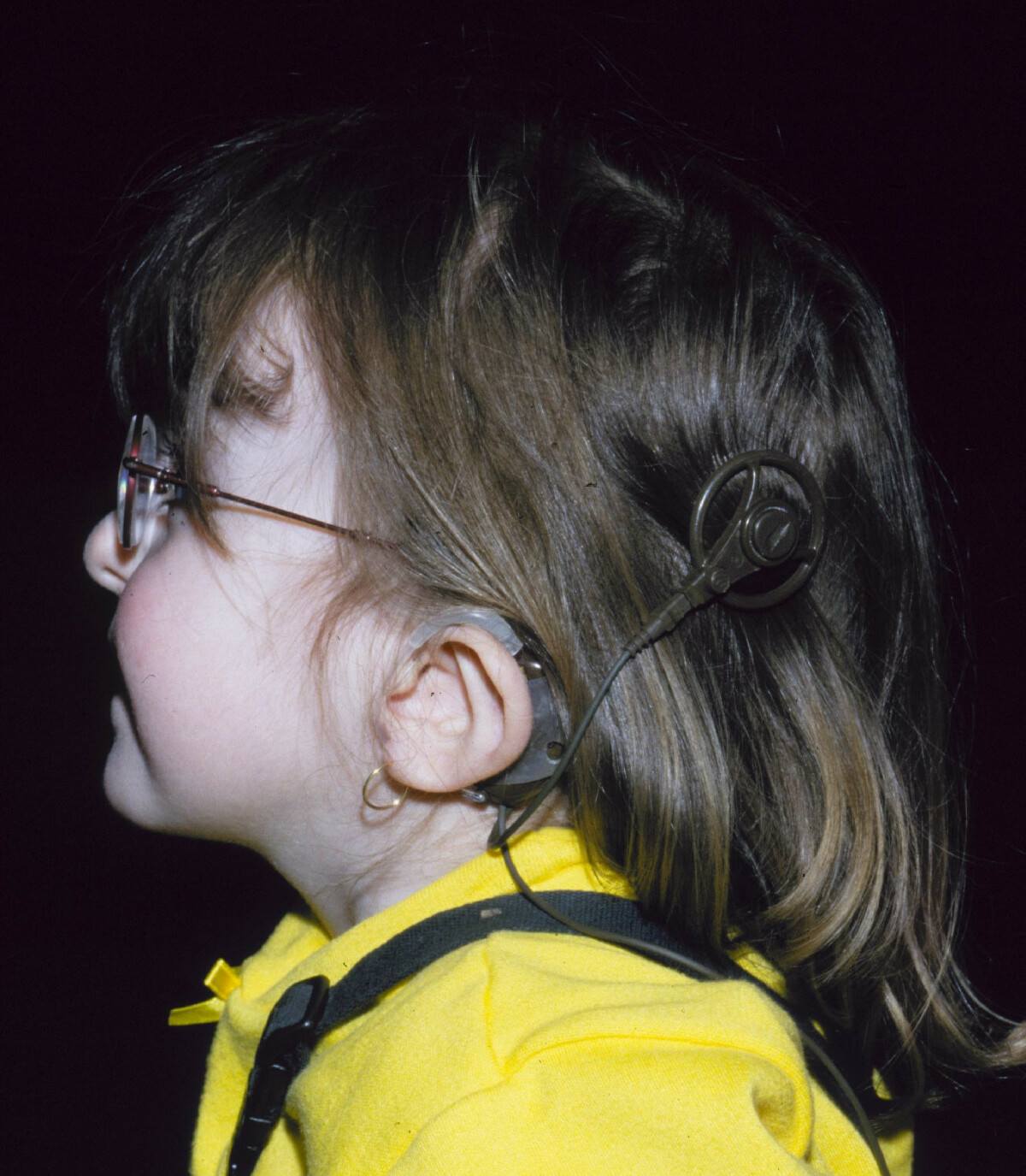
This article needs additional citations for verification . Please help improve this article by adding citations to reliable sources . Unsourced material may be challenged and removed. Find sources: "CHARGE syndrome" – news · newspapers · books · scholar · JSTOR ( July 2008 ) ( Learn how and when to remove this template message ) CHARGE syndrome "Lop ear" phenotype characteristic of a person with CHARGE syndrome, along with her cochlear implant . Specialty Medical genetics CHARGE syndrome (formerly known as CHARGE association ) is a rare syndrome caused by a genetic disorder . First described in 1979, the acronym "CHARGE" came into use for newborn children with the congenital features of coloboma of the eye, heart defects, atresia of the nasal choanae , retardation of growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness. [1] These features are no longer used in making a diagnosis of CHARGE syndrome, but the name remains. About two thirds of cases are due to a CHD7 mutation. CHARGE syndrome occurs only in 0.1–1.2 per 10,000 live births; as of 2009 it was the leading cause of congenital deafblindness in the US. [2] Contents 1 Genetics 2 Diagnosis 2.1 Signs 2.2 Genetic testing 2.3 Screening other organ systems 3 Therapy 3.1 Education 4 Epidemiology 5 History 6 References 7 External links Genetics [ edit ] CHARGE syndrome was formerly referred to as CHARGE association, which indicates a non-random pattern of congenital anomalies that occurs together more frequently than one would expect on the basis of chance.
CHARGE syndrome is a multiple congenital anomaly syndrome characterized by the variable combination of multiple anomalies, mainly Coloboma; Choanal atresia/stenosis; Cranial nerve dysfunction; Characteristic ear anomalies (known as the major 4 C's). Epidemiology The incidence is estimated to be 1/12,000 - 15,000 live births. Clinical description The syndrome shows a variable clinical picture, even within a family, depending on the associated anomalies. It presents in the neonatal period with cyanosis due to choanal atresia (60-70%, bony/membranous, unilateral/bilateral) and/or cyanotic heart disease (75-80%; e.g. conotruncal heart malformations, aortic arch defects; see these terms). Coloboma, more likely retinal, is present in 75-90% and can be in conjunction with microphthalmia and lead to vision loss.
CHARGE syndrome is a disorder that affects many areas of the body. CHARGE is an abbreviation for several of the features common in the disorder: coloboma, heart defects, atresia choanae (also known as choanal atresia), growth retardation, genital abnormalities, and ear abnormalities. The pattern of malformations varies among individuals with this disorder, and the multiple health problems can be life-threatening in infancy. Affected individuals usually have several major characteristics or a combination of major and minor characteristics. The major characteristics of CHARGE syndrome are common in this disorder and occur less frequently in other disorders. Most individuals with CHARGE syndrome have a gap or hole in one of the structures of the eye (coloboma), which forms during early development.
CHARGE syndrome is a congenital condition (present from birth) that affects many areas of the body. CHARGE stands for c oloboma, h eart defect, a tresia c hoanae (also known as choanal atresia ), r estricted growth and development, g enital abnormality, and e ar abnormality. Signs and symptoms vary among people with this condition; however, infants often have multiple life-threatening medical conditions. The diagnosis of CHARGE syndrome is based on a combination of major and minor characteristics. In more than half of all cases, mutations in the CHD7 gene cause CHARGE syndrome.
A form of von Willebrand disease (VWD) characterized by a bleeding disorder associated with a partial, quantitative plasmatic deficiency of an otherwise structurally and functionally normal von Willebrand factor (VWF). Epidemiology The type 1 disease is considered to be the most common form of VWD, accounting for between 50-75% of cases but its prevalence is probably overestimated. Clinical description Age of onset of bleeding anomalies varies, with earlier onset and more severe symptoms being associated with more severe VWF deficiency. The bleeding anomalies are generally characterized by mucocutaneous hemorrhage (menorrhagia, epistaxis, or prolonged bleeding after trauma or a surgical intervention). Hematomas and hemarthrosis are very rare. Etiology VWD is caused by mutations in the VWF gene (12p13.3).
Von Willebrand disease is a bleeding disorder that slows the blood clotting process , causing prolonged bleeding after an injury. People with this condition often experience easy bruising, long-lasting nosebleeds, and excessive bleeding or oozing following an injury, surgery, or dental work. Mild forms of von Willebrand disease may become apparent only when abnormal bleeding occurs following surgery or a serious injury. Women with this condition typically have heavy or prolonged bleeding during menstruation (menorrhagia), and some may also experience reproductive tract bleeding during pregnancy and childbirth. In severe cases of von Willebrand disease, heavy bleeding occurs after minor trauma or even in the absence of injury (spontaneous bleeding).
Munoz et al. (1988) described a brother and sister without a history of phacomatosis or cerebral tumors who developed malignant tumors with ependymal and choroidal differentiation. The girl presented at 28 months with a tumor of the posterior fossa, and the boy presented at 15 months with a tumor of the left cerebral hemisphere.
A complex group of benign and malignant cerebral tumors arising at any age. Epidemiology They are the most frequent cerebral tumors and represent more than half of all primary brain tumors. Incidence is estimated at 1/12,500. Clinical description The most frequent benign tumors are juvenile pilocytic astrocytomas (grade I) and diffuse low grade or fibrillary astrocytomas (grade II). The most frequent malignant tumors include anaplastic astrocytomas (grade III), glioblastomas (grade IV, the most severe form of astrocytoma; see this term), giant cell glioblastomas and gliosarcomas. Pleomorphic xanthoastrocytomas can be malignant or benign. These tumors occur at all ages although glioblastomas are more frequent in adults and in elderly people, while pilocytic astrocytomas are more frequent in children and adolescents.
This article includes a list of general references , but it remains largely unverified because it lacks sufficient corresponding inline citations . Please help to improve this article by introducing more precise citations. ( October 2014 ) ( Learn how and when to remove this template message ) Astrocytoma Two PET images — the upper of which shows a normal brain and the lower shows astrocytoma. Specialty Oncology , neurosurgery Astrocytomas are a type of brain tumor . They originate in a particular kind of glial cells, star-shaped brain cells in the cerebrum called astrocytes . This type of tumor does not usually spread outside the brain and spinal cord and it does not usually affect other organs.
Pursuant to the report by Berrettini et al. (1994) of a BPAD susceptibility locus on chromosome 18 and the report of a parent-of-origin effect by McMahon et al. (1995), Stine et al. (1995) undertook a linkage study in 28 nuclear families selected for apparent unilineal transmission of the BPAD phenotype.
Bipolar disorder is a mental health condition that causes extreme shifts in mood, energy, and behavior. This disorder most often appears in late adolescence or early adulthood, although symptoms can begin at any time of life. People with bipolar disorder experience both dramatic "highs," called manic episodes, and "lows," called depressive episodes. These episodes can last from hours to weeks, and many people have no symptoms between episodes. Manic episodes are characterized by increased energy and activity, irritability, restlessness, an inability to sleep, and reckless behavior.
This photo-activated signal transduction process is essential for vision. [13] The structure and function of rhodopsin and the gene encoding it have been the subjects of intense scrutiny for many years because rhodopsin serves as a useful model for understanding the largest receptor family in the human genome, the G protein-coupled receptors , and because defects in the rhodopsin gene are the most common cause of the most common inherited blinding disease, retinitis pigmentosa. [19] [28] [29] Rhodopsin Mutation [ edit ] The human rhodopsin gene is the locus for numerous alleles linked to the neurodegenerative disease retinitis pigmentosa. [19] Mutations in the rhodopsin gene account for 25% to 30% (30% to 40% according to [9] ) of all cases of autosomal dominant retinitis pigmentosa (ADRP). [10] [11] [12] [13] [20] Over 100 distinct mutations in the light-sensing molecule rhodopsin are known to cause (adRP). [7] [9] [13] [27] [30] Most of these mutations are missense mutations affecting single amino acid residues in the rhodopsin protein. [13] [31] These mutations affect rhodopsin transport to the outer segments of rod photoreceptor cells, rhodopsin folding, and rhodopsin endocytosis.
In people with incomplete injury, some or all of the spinal tracts involved in sexual responses remain intact, allowing, for example, orgasms like those of uninjured people. [22] In men, having an incomplete injury improves chances of being able to achieve erections [21] [23] and orgasms over those with complete injuries. [24] [25] Even people with complete SCI, in whom the spinal cord cannot transmit any messages past the level of the lesion, can achieve orgasm. [15] [17] [26] In 1960, in one of the earliest studies to look at orgasm and SCI, the term phantom orgasm was coined to describe women's perception of orgasmic sensations despite SCI—but subsequent studies have suggested the experience is not merely psychological. [10] Men with complete SCI report sexual sensations at the time of ejaculation, accompanied by physical signs normally found at orgasm, such as increased blood pressure. [26] Women can experience orgasm with vibration to the cervix regardless of level or completeness of injury; the sensation is the same as uninjured women experience. [27] The peripheral nerves of the parasympathetic nervous system that carry messages to the brain ( afferent nerve fibers ) may explain why people with complete SCI feel sexual and climactic sensations. [26] One proposed explanation for orgasm in women despite complete SCI is that the vagus nerve bypasses the spinal cord and carries sensory information from the genitals directly to the brain. [10] [25] [28] [29] Women with complete injuries can achieve sexual arousal and orgasm through stimulation of the clitoris, cervix, or vagina, which are each innervated by different nerve pathways, which suggests that even if SCI interferes with one area, function might be preserved in others. [30] In both injured and uninjured people, the brain is responsible for the way sensations of climax are perceived: the qualitative experiences associated with climax are modulated by the brain, rather than a specific area of the body. [26] Level of injury [ edit ] The effects of injury depend on the level along the spinal column .
Occupational exposure of pesticide factory workers to fenvalerate is associated with increased spermatozoa DNA damage. [28] Exposure to fenvalerate raised sex chromosome disomy 1.9-fold and disomy of chromosome 18 by 2.6-fold. [29] Exposure of male workers to carbaryl increased DNA fragmentation in spermatozoa, and also increased sex chromosome disomy by 1.7-fold and chromosome 18 disomy by 2.2-fold. [30] Humans are exposed to perfluorinated compounds (PFCs) in many commercial products. [31] Men contaminated with PFCs in whole blood or seminal plasma have spermatozoa with increased levels of DNA fragmentation and chromosomal aneuploidies. [31] Diagnosis [ edit ] Further information: Prenatal testing Example of Trisomy 21 detected via quantitative PCR short tandem repeat assay Germline aneuploidy is typically detected through karyotyping , a process in which a sample of cells is fixed and stained to create the typical light and dark chromosomal banding pattern and a picture of the chromosomes is analyzed.
Positron emission tomography (PET) scans of [18F]dopamine uptake showed a significant 28% increase in putamen dopamine storage after 18 months, suggesting a direct effect of GDNF on dopamine function. ... One or 2 mutant GBA alleles were found in 31 patients with Parkinson disease (31.3%): 28 were heterozygous and 3 were homozygous for one of these mutations.
Even small perturbances of proteins, such as the reduction of antithrombin to only 70–80% of the normal level, can increase the thrombosis risk; this is in contrast with hemophilia , which only arises if levels of coagulation factors are markedly decreased. [12] In addition to its effects on thrombosis, hypercoagulable states may accelerate the development of atherosclerosis , the arterial disease that underlies myocardial infarction and other forms of cardiovascular disease. [28] [29] Diagnosis [ edit ] A mutation of coagulation factor V (schematic representation drawn here) is much more common in people with thrombosis than in those without, but is only regarded as a weak risk factor.
A number sign (#) is used with this entry because susceptibility to thrombophilia (THPH1) can be conferred by heterozygous mutation in the thrombin gene (F2; 176930) on chromosome 11p11. Description Thrombophilia is a multifactorial disorder of inappropriate clot formation resulting from an interaction of genetic, acquired, and circumstantial predisposing factors. Venous thromboembolism most commonly manifests as deep vein thrombosis, which may progress to pulmonary embolism if the clot dislodges and travels to the lung. Other manifestations include thromboses of the cerebral or visceral veins and recurrent pregnancy loss (summary by Seligsohn and Lubetsky, 2001 and Varga and Kujovich, 2012). Genetic Heterogeneity of Thrombophilia THPH2 (188055) is caused by mutation in the F5 gene (612309) on chromosome 1q23; THPH3 (176860) and THPH4 (612304) are both caused by mutation in the PROC gene (612283) on 2q; THPH5 (612336) and THPH6 (614514) are caused by mutation in the PROS1 gene (176880) on 3q11; THPH7 (613118) is caused by mutation in the AT3 gene (107300) on 1q25; THPH8 (300807) is caused by mutation in the F9 gene (300746) on Xq27; THPH9 (612348) is associated with decreased release of tissue plasminogen activator (PLAT; 173370); THPH10 (612356) is caused by mutation in the HCF2 gene (142360) on 22q11; THPH11 (613116) is caused by mutation in the HRG gene (142640) on 3q27; and THPH12 (614486) is associated with variation in the THBD gene (188040) on 20p11.
Prothrombin-related thrombophilia affects the way the blood clots. Individuals who have a thrombophilia have an increased risk to form abnormal blood clots in blood vessels. Symptoms of prothrombin-related thrombophilia include a higher than average risk to develop blood clots in the deep veins of the legs ( deep venous thrombosis ) and blood clots in the lungs ( pulmonary embolism ). Many people with prothrombin-related thrombophilia never develop abnormal blood clots. This condition is caused by a particular genetic variant (written G20210A or 20210G>A) in the F2 gene and is inherited in an autosomal dominant pattern. Prothrombin-related thrombophilia is diagnosed based on the symptoms, physical exam, blood tests, and imaging studies.
Most of these lesions involved all cerebral lobes without hemispheric prevalence, but 28% of the myotonic dystrophy patients also showed particular white matter hyperintense lesions at their temporal poles.
Myotonic dystrophy type 1 (MD1), one of the two types of myotonic dystrophy , is an inherited type of muscular dystrophy that affects the muscles and other body systems (e.g., heart, eyes, endocrine system , and central nervous system). MD1 has three forms that somewhat overlap: the mild form, classic form, and congenital form (present at birth). The mild form has the least severe symptoms of the different forms of MD1 and is associated with a normal life span. The classic form is characterized by muscle weakness and wasting, prolonged muscle tensing ( myotonia ), cataract, and often, abnormal heart function. Adults with the classic form may become physically disabled and may have a shortened life span.
Summary Clinical characteristics. Myotonic dystrophy type 1 (DM1) is a multisystem disorder that affects skeletal and smooth muscle as well as the eye, heart, endocrine system, and central nervous system. The clinical findings, which span a continuum from mild to severe, have been categorized into three somewhat overlapping phenotypes: mild, classic, and congenital. Mild DM1 is characterized by cataract and mild myotonia (sustained muscle contraction); life span is normal. Classic DM1 is characterized by muscle weakness and wasting, myotonia, cataract, and often cardiac conduction abnormalities; adults may become physically disabled and may have a shortened life span. Congenital DM1 is characterized by hypotonia and severe generalized weakness at birth, often with respiratory insufficiency and early death; intellectual disability is common.
Myotonic dystrophy is part of a group of inherited disorders called muscular dystrophies. It is the most common form of muscular dystrophy that begins in adulthood. Myotonic dystrophy is characterized by progressive muscle wasting and weakness. People with this disorder often have prolonged muscle contractions (myotonia) and are not able to relax certain muscles after use. For example, a person may have difficulty releasing their grip on a doorknob or handle.
A rare genetic multi-system disorder characterized by a wide range of muscle-related manifestations (muscle weakness, myotonia, early onset cataracts (before age 50) and systemic manifestations (cerebral, endocrine, cardiac, gastrointestinal tract, uterus, skin and immunologic involvement) that vary depending on the age of onset. The very wide clinical spectrum ranges from lethal presentations in infancy to mild, late-onset disease. Epidemiology It is the most frequent adult muscular dystrophy and has an estimated prevalence ranging from 1/215,000 in Taiwan to 1/5,500 in Croatia. It appears to be more prevalent in the Saguenay-Lac-St-Jean region-Quebec, Canada (1/600), suggesting a founder effect. The disease occurs worldwide. Clinical description The age of onset is highly variable, from prenatal to adulthood.