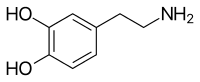
The authors noted the phenotypic similarities to Renpenning syndrome/MRXS8 (RENS1; 309500). Shrimpton et al. (2000) narrowed the critical region to an 11-cM (22-Mb) interval between DXS8040 and DXS1217, increasing the lod score to 4.75.
Summary The purpose of this overview is to increase the awareness of clinicians regarding genetic causes of nuclear gene-encoded Leigh syndrome spectrum (LSS), management, especially treatable disorders, and relevant genetic counseling issues.
A number sign (#) is used with this entry because multiple congenital anomalies-hypotonia-seizures syndrome-2 (MCAHS2) is caused by mutation in the PIGA gene (311770) on chromosome Xp22. ... The family was previously reported as having West syndrome by Claes et al. (1997) (family B). ... One of the patients had been diagnosed clinically with Schinzel-Giedion syndrome (269150) (Watanabe et al., 2012). ... Fauth et al. (2016) reported 3 male patients from 2 unrelated families with a lethal multiple congenital anomaly syndrome. One died at age 15 days, 1 died at age 3 months, and the third died in utero. ... Molecular Genetics By exome sequencing of the X chromosome in a family with multiple congenital anomalies-hypotonia-seizures syndrome-2, Johnston et al. (2012) identified a germline mutation in the PIGA gene (R412X; 311770.0011).
Multiple congenital anomalies-hypotonia-seizures syndrome type 2 (MCAHS2) is a genetic neurodevelopmental disorder characterized by distinctive facial features, low muscle tone ( hypotonia ) at birth, myoclonic seizures (which cause jerks or twitches of the upper body, arms, or legs), and various other problems involving the central nervous system, heart, and urinary system.
A rare, life-threatening, congenital, non-syndromic, conotruncal heart malformation disease characterized by absent or severely undeveloped pulmonary valve leaflets (with a restrictive ring of thickened tissue at the place of the pulmonary valve annulus), associated with an intact ventricular septum and a patent ductus arteriosus, manifesting with marked respiratory insufficiency.
Encephalopathy-hypertrophic cardiomyopathy-renal tubular disease syndrome is a rare mitochondrial disease due to a defect in coenzyme Q10 biosynthesis that manifests with a broad spectrum of signs and symptoms which may include: neonatal lactic acidosis, global developmental delay, tonus disorder, seizures, reduced spontaneous movements, ventricular hypertrophy, bradycardia, renal tubular dysfunction with massive lactic acid excretion in urine, severe biochemical defect of respiratory chain complexes II/III when assayed together and deficiency of coenzyme Q10 in skeletal muscle.
Cranial ultrasound showed multiple choroid plexus cysts and abnormal signals in the basal ganglia, suggesting a neonatal Leigh-like syndrome. He had reduced spontaneous movements with intermittent opisthotonus, seizures, and recurrent episodes of apnea, and died at 18 days of age.
Global developmental delay-visual anomalies-progressive cerebellar atrophy-truncal hypotonia syndrome is a rare, genetic, neurological disorder characterized by mild to severe developmental delay and speech impairment, truncal hypotonia, abnormalities of vision (including cortical visual impairment and abnormal visual-evoked potentials), progressive brain atrophy mainly affecting the cerebellum, and shortened or atrophic corpus callosum.
A number sign (#) is used with this entry because of evidence that cerebellar atrophy, visual impairment, and psychomotor retardation (CAVIPMR) is caused by homozygous mutation in the EMC1 gene (616846) on chromosome 1p36. Clinical Features Harel et al. (2016) reported 6 children from 3 unrelated families of different ethnic origin with a severe neurodegenerative disorder apparent since birth. The children, ranging in age from 3 to 14 years, had global developmental delay, speech delay, and hypotonia associated with cerebellar atrophy and a short or atrophic corpus callosum. Four patients from 2 families had a more severe disorder, with profound intellectual disability, progressive microcephaly (up to -4 SD), increased tone in the extremities, hyporeflexia, dystonic posturing, scoliosis, and cerebral atrophy. All patients had some variable dysmorphic features, including deep-set eyes, gingival hyperplasia, retrognathia, and short philtrum.
The newly described 2q23.1 microdeletion syndrome includes severe intellectual deficit with pronounced speech delay, behavioral abnormalities including hyperactivity and inappropriate laughter, short stature and seizures.
MBD5 -associated neurodevelopmental disorder (MAND) is a condition that affects neurological and physical development. Children with MAND have mild to severe intellectual disability and developmental delay. They often have poor coordination and do not walk until age 2 or 3. Their walking style (gait) is often unbalanced and wide-based. Language skills, both the production of speech and the ability to understand speech, are very limited in affected individuals. By age 2, most children with MAND develop recurring seizures (epilepsy).
MBD25–related intellectual disability, or MBD25 haploinsufficiency, is a neurological and developmental disorder characterized by developmental delay, intellectual disability , speech problems, seizures, sleep troubles, and abnormal behaviors. Most children lack speech entirely or may only be able to use single words, short phrases, or short sentences. Seizures are present in about 80% and usually begin around age two years. Sleep troubles, present in about 80% of children, can cause daytime drowsiness. Abnormal behaviors can include autistic-like-behavior (80%) and self-injury and aggression (60%).
These features led to the initial clinical impression of Angelman syndrome (105830), Rett syndrome (RTT; 312750), or Smith-Magenis syndrome (SMS; 182290) in several patients. ... Inheritance Among 11 patients with chromosome 2q23.1 deletion syndrome, van Bon et al. (2010) found that all with available parent samples had a de novo deletion. ... In a review of 15 patients with chromosome 2q23.1 deletion syndrome, van Bon et al. (2010) found that the deletion sizes ranged from 250 kb to 5.5 Mb comprising 15 genes. ... In a patient with some features of Kleefstra syndrome (610253), Kleefstra et al. (2012) detected a frameshift mutation in the MBD5 gene (611472.0003). The patient was 1 of 9 patients with syndromic mental retardation who shared core features of Kleefstra syndrome but were phenotypically heterogeneous otherwise, with a phenotype referred to by the authors as Kleefstra syndrome spectrum (KSS) .
A number sign (#) is used with this entry because of evidence that intellectual developmental disorder with dysmorphic facies, seizures, and distal limb anomalies (IDDFSDA) is caused by homozygous mutation in the OTUD6B gene (612021) on chromosome 8q21. Description IDDFSDA is an autosomal recessive severe multisystem disorder characterized by poor overall growth, developmental delay, early-onset seizures, intellectual disability, and dysmorphic features. There is phenotypic variability. The most severely affected patients have a neurodevelopmental disorder with microcephaly, absent speech, and inability to walk, and they require feeding tubes. Some patients have congenital heart defects or nonspecific abnormalities on brain imaging. Less severely affected individuals have mild to moderate intellectual disability with normal speech and motor development (summary by Santiago-Sim et al., 2017).
A rare genetic multiple congenital anomalies/dysmorphic syndrome characterized by developmental delay with mild intellectual disability, short stature, facial dysmorphism (such as sparse hair, high forehead, deep-set eyes, short and upslanting palpebral fissures, short nose, anteverted nares, wide nasal base with broad nasal tip and broad columella, long philtrum, thin upper lip, and low-set, posteriorly rotated ears), and variable onset of sensorineural hearing loss and retinitis pigmentosa.
A number sign (#) is used with this entry because of evidence that short stature, hearing loss, retinitis pigmentosa, and distinctive facies (SHRF) is caused by homozygous or compound heterozygous mutation in the EXOSC2 gene (602238) on chromosome 9q34. Description SHRF is an autosomal recessive disorder characterized by short stature, brachydactyly, dysmorphic facial features, hearing loss, and visual impairment. Onset of the hearing and visual abnormalities, including retinitis pigmentosa, varies from birth to the second decade. Patients have mild intellectual disability and mild cerebellar atrophy with myelination defects on brain imaging (summary by Di Donato et al., 2016). Clinical Features Di Donato et al. (2016) reported 3 patients from 2 unrelated German families with a similar complex congenital phenotype.
Chromosome 1q41-q42 deletion syndrome is characterized by a small, but variable deletion in a particular place on the long arm of one copy of chromosome 1 , usually spanning several genes.
Ovarian remnant syndrome (ORS) is characterized by the presence of residual ovarian tissue after a woman has had surgery to remove one ovary or both ovaries ( oophorectomy ).
Ovarian remnant syndrome Other names Residual ovary syndrome Ovarian remnant syndrome [1] is a condition that occurs when ovarian tissue is left behind following oophorectomy , causing development of a pelvic mass, pelvic pain , and occasionally dyspareunia . [2] Ovarian remnant syndrome (ORS) is characterized by the presence of residual ovarian tissue after a woman has had surgery to remove one ovary or both ovaries ( oophorectomy ). [3] Contents 1 Signs and symptoms 2 Cause 2.1 Risks 3 Diagnosis 4 Treatment 5 Epidemiology 6 References 7 External links Signs and symptoms [ edit ] If ovarian hormones are present after the ovaries are removed can be a sign that ovarian tissue still remains. [4] Signs and symptoms may include pelvic pain, a pelvic mass, or the absence of menopause after oophorectomy. ... Hormonal therapy to suppress ovarian function is an alternative treatment for those who refuse surgery, or those who are not candidates for surgery. [3] Medications may be used to treat ORS and include GnRH agonists, danazol, or progesterone. [4] Epidemiology [ edit ] The incidence of ovarian remnant syndrome is difficult to determine. [3] [4] The available data are limited to case reports or to retrospective case series. ... Steege, MD. "What is ovarian remnant syndrome?" . HealthyWomen . National Women's Health Resource Center, Inc . ... ISBN 9780803629790 . ^ a b c d e f g h "Ovarian remnant syndrome" . rarediseases.info.nih.gov . ... External links [ edit ] Classification D ICD - 10 : N99.8 ICD - 9-CM : 629 Ovarian remnant syndrome v t e Female reproductive system Internal Adnexa Ovaries Follicles corpus hemorrhagicum luteum albicans Theca of follicle externa interna Follicular antrum Follicular fluid Corona radiata Zona pellucida Membrana granulosa Perivitelline space Other Germinal epithelium Tunica albuginea cortex Cumulus oophorus Stroma Medulla Fallopian tubes Isthmus Ampulla Infundibulum Fimbria Ostium Ligaments Ovarian ligament Suspensory ligament Wolffian vestiges Gartner's duct Epoophoron Vesicular appendages of epoophoron Paroophoron Uterus Regions Body Uterine cavity Fundus Cervix External orifice Cervical canal Internal orifice Supravaginal portion Vaginal portion Uterine horns Layers Endometrium epithelium Myometrium Perimetrium Parametrium Ligaments Round ligament Broad ligament Cardinal ligament Uterosacral ligament Pubocervical ligament General Uterine glands Vagina Fossa of vestibule of vagina Vaginal fornix Hymen Vaginal rugae Support structures Vaginal epithelium External Vulva Labia Mons pubis Labia majora Anterior commissure Posterior commissure Pudendal cleft Labia minora Frenulum of labia minora Frenulum of clitoris Vulval vestibule Interlabial sulci Bulb of vestibule Vaginal orifice vestibular glands/ducts Bartholin's glands/Bartholin's ducts Skene's glands/Skene's ducts Clitoris Crus of clitoris Body of clitoris ( Corpus cavernosum ) Clitoral glans Hood Urethra Urethral crest Other G-spot Urethral sponge Perineal sponge
Although the underlying genetic cause of some hereditary cases is unknown, many are part of a hereditary cancer syndrome (such as BRCA1 or BRCA2 hereditary breast and ovarian cancer syndrome , Lynch syndrome and Peutz-Jeghers syndrome ) and are inherited in an autosomal dominant manner.
Familial ovarian cancer may be part of other cancer syndromes. See susceptibility to familial breast-ovarian cancer 1 and 2 (604370 and 612555), due to mutations in the BRCA1 (113705) and BRCA2 (600185) genes, respectively; and Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer (see, e.g., HNPCC1; 120435), due to mutations in DNA mismatch repair genes such as MSH2 (609309), MSH3 (600887), MSH6 (600678), and MLH1 (120436). ... Some of these families may have had the breast-ovarian cancer syndrome or Lynch syndrome. Liber (1950) described a family with histologically proven papillary adenocarcinoma of the ovary in 5 sisters and their mother. ... In addition to colon cancer, ovarian cancer may be a manifestation of this syndrome. No germline mutations were identified in any of the other genes analyzed, including BRCA1, the 'ovarian cancer-cluster region' (nucleotides 3139-7069) of BRCA2, and MSH2. ... Liede et al. (1998) concluded that site-specific ovarian cancer families probably represent a variant of the breast-ovarian cancer syndrome, attributable to mutation in either BRCA1 or BRCA2. ... About 50% of the women developed another cancer in the HNPCC/Lynch syndrome tumor spectrum. The 5-, 10-, 20-, and 30-year survival specific for deaths due to HNPCC/Lynch syndrome-associated cancers were 79.2%, 75.7%, 68.4% and 47.3%, respectively.
Malouf et al. (1985) suggested that males with this syndrome may have cardiomyopathy but not testicular dysgenesis. Harbord et al. (1989) described cardiomyopathy in association with Martsolf syndrome (212720), which has hypogonadism as a feature. ... Nguyen et al. (2007) stated that the features of this patient were consistent with atypical Werner syndrome (see 277700). McPherson et al. (2009) restudied the patient originally described by Nguyen et al. (2007) and reported additional features, including premature ovarian failure with secondary amenorrhea at 15 years of age. ... Although Chen et al. (2003) designated the patient as having 'atypical Werner syndrome' (see 277700), Hegele (2003) suggested that the patient more likely had late-onset Hutchinson-Gilford progeria syndrome (see 176670). ... In a 17-year-old Caucasian female with premature ovarian failure and dilated cardiomyopathy, who had features consistent with atypical Werner syndrome (see 277700) but who was negative for mutation in the RECQL2 gene (604611), Nguyen et al. (2007) identified heterozygosity for a missense mutation in the LMNA gene (L59R; 150330.0052).
Dilated cardiomyopathy with hypergonadotropic hypogonadism (DCMHH) is a condition that primarily affects the heart and gonads (male testes or female ovaries). It is characterized by a disease of the heart muscle ( dilated cardiomyopathy ) and little or no production of sex hormones due to a problem with the pituitary gland or hypothalamus ( hypergonadotropic hypogonadism ). Other symptoms might include: characteristic facial features, intellectual disability, mild skeletal anomalies, and abnormalities of the metabolic system. Some cases of DCMHH are caused by mutations in the LMNA gene. Both autosomal dominant and autosomal recessive inheritance patterns have been described. Although there is no specific treatment or cure for DCMHH, there are ways to manage the symptoms.
Fiskerstrand type peripheral neuropathy is a slowly-progressive Refsum-like disorder associating signs of peripheral neuropathy with late-onset hearing loss, cataract and pigmentary retinopathy that become evident during the third decade of life. Epidemiology The syndrome has been described in three patients from a consanguineous family (one brother, one sister and a male cousin).
A number sign (#) is used with this entry because polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract (PHARC) is caused by homozygous or compound heterozygous mutation in the ABHD12 gene (613599) on chromosome 20p11. Clinical Features Fiskerstrand et al. (2009) reported a consanguineous Norwegian family in which 3 individuals had a slowly progressive neurologic disorder resembling the clinical features of Refsum disease (266500). The authors suggested naming the disorder PHARC, an acronym that describes the major features of the disorder. Features in childhood included pes cavus and Achilles tendon contractures; 1 had poor hearing in childhood. Hearing loss and visual problems related to cataracts developed in the third decade.
In this condition, the difference between pupils is usually less than 1 mm. [3] Horner's syndrome Mechanical anisocoria : Occasionally previous trauma, eye surgery, or inflammation ( uveitis , angle closure glaucoma ) can lead to adhesions between the iris and the lens. ... It may be associated with loss of deep tendon reflex (Adie's syndrome). Tonic pupil is characterized by delayed dilation of iris especially after near stimulus, segmental iris constriction, and sensitivity of pupil to a weak solution of pilocarpine. ... Anisocoria which is worsened (greater asymmetry between the pupils) in the dark suggests the small pupil (which should dilate in dark conditions) is the abnormal pupil and suggests Horner's syndrome or mechanical anisocoria. In Horner's syndrome sympathetic nerve fibers have a defect, therefore the pupil of the involved eye will not dilate in darkness. ... Some of the causes of anisocoria are life-threatening, including Horner's syndrome (which may be due to carotid artery dissection ) and oculomotor nerve palsy (due to a brain aneurysm, uncal herniation , or head trauma). ... See also [ edit ] Cycloplegia Miosis Mydriasis Parinaud's syndrome References [ edit ] ^ Lam, BL; Thompson, HS; Corbett, JJ (Jul 15, 1987).
Unequal pupil size without associated features of the Horner syndrome (143000) or any other abnormality has been observed in a dominant pedigree pattern.
Dopamine dysregulation syndrome Two-dimensional skeletal formula of the dopamine molecule. ... The behavioral and mood symptoms of the syndrome are produced by the dopamine overdose . [4] Diagnosis [ edit ] Diagnosis of the syndrome is clinical since there are no laboratory tests to confirm it. ... "Dopamine dysregulation syndrome, addiction and behavioral changes in Parkinson's disease". ... "Resolution of dopamine dysregulation syndrome following cessation of dopamine agonist therapy in Parkinson's disease". ... "Valproate as a treatment for dopamine dysregulation syndrome (DDS) in Parkinson's disease".
Eosinophilic fasciitis is a very rare condition in which muscle tissue underneath the skin, called fascia, becomes swollen and thick. Rapid swelling can occur in the hands, arms, legs, and feet. People with this condition have a buildup of eosinophils, a type of white blood cell, in the affected fascia and muscles. The exact cause of this condition is unknown. Corticosteroids and other immune-suppressing medications are used to relieve the symptoms. Eosinophilic fasciitis is similar in appearance to scleroderma . However, in contrast with systemic sclerosis, internal organ involvement in eosinophilic fasciitis is generally absent. Some researchers believe that eosinophilic fasciitis may be a variant of morphea (localized scleroderma).
A rare idiopathic inflammatory myopathy that is characterized by inflammation and thickening of the fascia, usually associated with peripheral eosinophilia. It presents during adulthood with symmetrical and painful swelling of mainly the extremities that progressively become indurated. Fatigue, disabling cutaneous fibrosis, myositis and arthritis may also be observed.
Thomson et al. (1989) described a brother and sister, aged 33 and 38 years, respectively, who developed eosinophilic fasciitis within a period of 6 months. They were found to have identical HLA-A, -B, -DR, and -DQ antigens. No common environmental factors close to the time of onset were identified. Muscle - Eosinophilic fasciitis Inheritance - Autosomal recessive ▲ Close