Lethal left ventricular non-compaction-seizures-hypotonia-cataract-developmental delay syndrome is rare, genetic, neurometabolic disease characterized by global developmental delay, severe hypotonia, seizures, cataracts, cardiomyopathy (including left or bi-ventricular hypertrophy, dilated cardiomyopathy) and left ventricular non-compaction, typically resulting in infantile or early-childhood death.
A number sign (#) is used with this entry because of evidence that combined oxidative phosphorylation deficiency-31 (COXPD31) is caused by homozygous or compound heterozygous mutation in the MIPEP gene (602241) on chromosome 13q12. Description Combined oxidative phosphorylation deficiency-31 is an autosomal recessive multisystem disorder characterized by left ventricular noncompaction (LVNC), global developmental delay, and severe hypotonia. More variable features include seizures, cataract, and abnormal movements. The disorder becomes apparent soon after birth or in early infancy, and patients may die in early childhood. Biochemical studies are consistent with a defect in mitochondrial function (summary by Eldomery et al., 2016).
Sudden infant death syndrome (SIDS) is the unexpected, sudden death of a child under age 1 which cannot be explained after a thorough investigation is conducted.
Description Sudden infant death syndrome (SIDS) is a diagnosis of exclusion which should be made only after a thorough autopsy without identification of a specific cause of death (Mage and Donner, 2004). ... Folgering et al. (1979) reported an infant with the congenital central hypoventilation syndrome (209880) and absence of the arcuate nucleus at autopsy. ... Although Schwartz et al. (1998) stated that they ruled out the long QT syndrome on the basis of family history, only 39% of all established LQTS cases have a positive family history (Garson et al., 1993). ... They found the same mutation in affected members of a family with long QT syndrome. Schwartz et al. (2001) concluded that these findings confirmed the hypothesis that some deaths from SIDS are caused by long QT syndrome and supported implementation of neonatal electrocardiographic screening during the second or third week of life when the risk of spuriously long QT intervals (false positives) is extremely small. ... Another well-studied example is long QT syndrome. By contrast, evidence tended to exclude hypoglycemia and thrombosis as major causes of SIDS.
"Maternal alcohol use and sudden infant death syndrome and infant mortality excluding SIDS". ... (May 2009). "Contribution of long-QT syndrome genetic variants in sudden infant death syndrome". ... "Distinguishing sudden infant death syndrome from child abuse fatalities" . ... "Immunisation and the sudden infant death syndrome. New Zealand Cot Death Study Group" . ... "A competing risk model of sudden infant death syndrome incidence in two US birth cohorts".
Overview Sudden infant death syndrome is the unexplained death of a baby. ... It often happens during sleep. Sudden infant death syndrome also is known as SIDS . It is sometimes called crib death because infants often die in their cribs.
In humans this may refer to: 45, X , also known as Turner syndrome 45,X/46,XY mosaicism 46, XX/XY 47, XXX , also known as Triple X syndrome and trisomy X 47, XXY , also known as Klinefelter syndrome 47, XYY , has normal phenotype 48, XXXX 48, XXXY 48, XXYY 49, XXXXY 49, XXXXX XX gonadal dysgenesis XY gonadal dysgenesis XX male syndrome Index of articles associated with the same name This article includes a list of related items that share the same name (or similar names).
PMID 24507822 . ^ "Paraneoplastic syndromes of the nervous system" . Mayo Clinic . ... "Sympathetic nervous system dysfunction in fibromyalgia, chronic fatigue syndrome, irritable bowel syndrome, and interstitial cystitis: a review of case-control studies" . ... "Postural orthostatic tachycardia syndrome (POTS): a diagnostic dilemma" . ... "Vegetative-Vascular Dystonia and Osteoalgetic Syndrome or Chronic Fatigue Syndrome as a Characteristic After-Effect of Radioecological Disaster". ... "[Thermography in healthy subjects and in the syndrome of vegetative-vascular dystonia]".
ISBN 978-0-19-856634-2 . v t e Diseases of the autonomic nervous system General Dysautonomia Autonomic dysreflexia Autonomic neuropathy Pure autonomic failure Hereditary Hereditary sensory and autonomic neuropathy Familial dysautonomia Congenital insensitivity to pain with anhidrosis Orthostatic intolerance Orthostatic hypotension Postural orthostatic tachycardia syndrome Other Horner's syndrome Multiple system atrophy
Tylosis with esophageal cancer Specialty Medical genetics Howel–Evans syndrome is an extremely rare condition involving thickening of the skin in the palms of the hands and the soles of the feet ( hyperkeratosis ). ... It was first described in 1958. [2] Contents 1 Presentation 2 Genetics 3 Molecular biology 3.1 Other associations 3.2 Related genes 4 Diagnosis 4.1 Differential diagnosis 5 Treatment 6 Terminology 7 See also 8 References 9 External links Presentation [ edit ] This condition is inherited as an autosomal dominant syndrome and characterized by palmoplantar keratoderma, oral precursor lesions particularly on the gums ( leukoplakia ) and a high lifetime risk of esophageal cancer (95% develop esophageal cancer by the age of 65). [3] Relapsing cutaneous horns of the lips has been reported in this condition. [4] There are several types of this condition have been described – epidermolytic (Vörner type) and non-epidermolytic. ... Other possible associations include corneal defects, congenital pulmonary stenosis , [19] total anomalous pulmonary venous connection [20] deafness [21] and optic atrophy . [22] Related genes [ edit ] A related gene – Rhomboid domain containing 2 ( RHBDD2 ) – appears to be important in breast cancer . [23] A second related gene – rhomboid family 1 ( RHBDF1 ) – appears to be important in head and neck cancer. [24] A third member of this family – RHBDD1 – cleaves Bcl-2-interacting killer ( BIK ) – a proapoptotic member of the B cell lymphoma 2 ( Bcl-2 ) family. [25] These proteins may also have a role in diabetes . [26] Diagnosis [ edit ] Differential diagnosis [ edit ] The differential diagnosis is quite extensive and includes [27] [28] Buschke–Fischer–Brauer disease Curth–Macklin ichthyosis Gamborg Nielsen syndrome Greither disease Haber syndrome Hereditary punctate palmoplantar keratoderma Jadassohn–Lewandowsky syndrome Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenital and sclerosing keratoderma syndrome Meleda disease Mucosa hyperkeratosis syndrome Naegeli–Franceschetti–Jadassohn syndrome Naxos disease Olmsted syndrome Palmoplantar keratoderma and leukokeratosis anogenitalis Pandysautonomia Papillomatosis of Gougerot and Carteaud Papillon–Lefèvre syndrome Punctate porokeratotic keratoderma Richner–Hanhart syndrome Schöpf–Schulz–Passarge syndrome Unna Thost disease Vohwinkel syndrome Wong's dermatomyositis Treatment [ edit ] Systemic retinoids are the drugs used for tylosis. ... Am J Gastroenterol 93(3):449–451 ^ Grundmann JU, Weisshaar E, Franke I, Bonnekoh B, Gollnick H (2003) Lung carcinoma with congenital plantar keratoderma as a variant of Clarke-Howel-Evans syndrome. Int J Dermatol 42(6):461–463 ^ Nomori H, Horio H, Iga R, Fuyuno G, Kobayashi R, Morinaga S (1996) Squamous cell carcinoma of the lung associated with palmo-plantar hyperkeratosis.
Description Palmoplantar keratoderma (PPK) is a complex group of hereditary syndromes that have been classified into diffuse, punctate, and focal forms according to the pattern of hyperkeratosis on the palms and soles (Lucker et al., 1994). ... Stevens et al. (1996) referred to the Howell-Evans syndrome as palmoplantar ectodermal dysplasia type III. ... Mapping In mapping the genetic locus for keratosis palmaris et plantaris (without associated features) to chromosome 17 near the keratin type I genes, Rogaev et al. (1993) raised the possibility that the association with cancer may represent a contiguous gene syndrome. Blanchet-Bardon et al. (1987) observed hereditary epidermolytic palmoplantar keratoderma in association with breast and ovarian cancer in a large kindred.
Tylosis with esophageal cancer (TOC) is an inherited condition that increases the risk for esophageal cancer. The symptoms of TOC include thickening of the skin on the palms and soles of the feet (palmoplantar keratoderma) and white lesions inside the mouth. People with TOC are at very high risk to develop esophageal cancer. The palmoplantar keratoderma usually occurs in childhood, and esophageal cancer usually occurs in adulthood. TOC is caused by a variant in the RHBDF2 gene and is inherited in an autosomal dominant pattern. Diagnosis is based on the symptoms, clinical exam, and family history.
A rare genetic disease characterized by thickening of the skin on palms and soles restricted to areas of weight bearing and/or friction (focal, non-epidermolytic palmoplantar keratoderma) and oral and esophageal leukokeratosis, associated with a very high lifetime risk of developing squamous cell carcinoma of the esophagus. The skin lesions appear in childhood and can be complicated by fissuring and infection.
Aortoiliac occlusive disease Other names Leriche's syndrome and Leriche syndrome Plate from Gray's Anatomy showing the abdominal aorta and the common iliac arteries. Specialty Cardiology Fluoroscopic image of an aorta affected by Leriche's syndrome In medicine , aortoiliac occlusive disease , is a form of central artery disease involving the blockage of the abdominal aorta as it transitions into the common iliac arteries . Contents 1 Signs and symptoms 2 Diagnosis 3 Treatment 4 History 5 See also 6 References 7 External links Signs and symptoms [ edit ] Classically, it is described in male patients as a triad of the following signs and symptoms : claudication of the buttocks and thighs absent or decreased femoral pulses erectile dysfunction This combination is known as Leriche syndrome. However, any number of symptoms may present, depending on the distribution and severity of the disease, such as muscle atrophy, slow wound healing in the legs, and critical limb ischemia . ... ^ Leriche, R; Morel, A (February 1948). "The Syndrome of Thrombotic Obliteration of the Aortic Bifurcation" . ... External links [ edit ] Classification D ICD - 9-CM : 444.0 MeSH : D007925 DiseasesDB : 29335 External resources eMedicine : med/2759 v t e Cardiovascular disease (vessels) Arteries , arterioles and capillaries Inflammation Arteritis Aortitis Buerger's disease Peripheral artery disease Arteriosclerosis Atherosclerosis Foam cell Fatty streak Atheroma Intermittent claudication Critical limb ischemia Monckeberg's arteriosclerosis Arteriolosclerosis Hyaline Hyperplastic Cholesterol LDL Oxycholesterol Trans fat Stenosis Carotid artery stenosis Renal artery stenosis Other Aortoiliac occlusive disease Degos disease Erythromelalgia Fibromuscular dysplasia Raynaud's phenomenon Aneurysm / dissection / pseudoaneurysm torso : Aortic aneurysm Abdominal aortic aneurysm Thoracic aortic aneurysm Aneurysm of sinus of Valsalva Aortic dissection Aortic rupture Coronary artery aneurysm head / neck Intracranial aneurysm Intracranial berry aneurysm Carotid artery dissection Vertebral artery dissection Familial aortic dissection Vascular malformation Arteriovenous fistula Arteriovenous malformation Telangiectasia Hereditary hemorrhagic telangiectasia Vascular nevus Cherry hemangioma Halo nevus Spider angioma Veins Inflammation Phlebitis Venous thrombosis / Thrombophlebitis primarily lower limb Deep vein thrombosis abdomen Hepatic veno-occlusive disease Budd–Chiari syndrome May–Thurner syndrome Portal vein thrombosis Renal vein thrombosis upper limb / torso Mondor's disease Paget–Schroetter disease head Cerebral venous sinus thrombosis Post-thrombotic syndrome Varicose veins Gastric varices Portacaval anastomosis Caput medusae Esophageal varices Hemorrhoid Varicocele Other Chronic venous insufficiency Chronic cerebrospinal venous insufficiency Superior vena cava syndrome Inferior vena cava syndrome Venous ulcer Arteries or veins Angiopathy Macroangiopathy Microangiopathy Embolism Pulmonary embolism Cholesterol embolism Paradoxical embolism Thrombosis Vasculitis Blood pressure Hypertension Hypertensive heart disease Hypertensive emergency Hypertensive nephropathy Essential hypertension Secondary hypertension Renovascular hypertension Benign hypertension Pulmonary hypertension Systolic hypertension White coat hypertension Hypotension Orthostatic hypotension
The incidence is 14 per 100,000 and the condition affects 3-11% of blind children. [ citation needed ] Causes [ edit ] Microphthalmia in newborns is sometimes associated with fetal alcohol syndrome [1] or infections during pregnancy, particularly herpes simplex virus , rubella and cytomegalovirus (CMV), but the evidence is inconclusive. Genetic causes of microphthalmia include chromosomal abnormalities ( Trisomy 13 ( Patau syndrome ), Triploid Syndrome , 13q deletion syndrome , and Wolf-Hirschhorn Syndrome ) or monogenetic Mendelian disorders. ... Without this fluid, the eye fails to enlarge, thus the name microphthalmia.The gene encoding the microphthalmia-associated transcription factor (MITF) is a member of the basic helix-loop-helix -leucine zipper (bHLH-ZIP) family. Waardenburg syndrome type 2 (WS type 2) in humans is also a type of microphthalmia syndrome. Mutations in MITF gene are thought to be responsible for this syndrome. The human MITF gene is homologous to the mouse MITF gene (aka mouse mi or microphthalmia gene); mouse with mutations in this gene are hypopigmented in their fur. ... Further reading [ edit ] GeneReviews/NCBI/NIH/UW entry on Anophthalmia / Microphthalmia Overview GeneReviews/NCBI/NIH/UW entry on Microphthalmia with Linear Skin Defects Syndrome OMIM-Online Mendelian Inheritance in Man External links [ edit ] Classification D ICD - 10 : Q11.2 ICD - 9-CM : 743.1 MeSH : D008850 DiseasesDB : 29618 External resources eMedicine : oph/572 GeneReviews : Anophthalmia / Microphthalmia Overview v t e Congenital malformations and deformations of eyes Adnexa Eyelid Ptosis Ectropion Entropion Distichia Blepharophimosis Ablepharon Marcus Gunn phenomenon Lacrimal apparatus Congenital lacrimal duct obstruction Globe Entire eye Anophthalmia ( Cystic eyeball , Cryptophthalmos ) Microphthalmia Lens Ectopia lentis Aphakia Iris Aniridia Anterior segment Axenfeld–Rieger syndrome Cornea Keratoglobus Megalocornea Other Buphthalmos Coloboma ( Coloboma of optic nerve ) Hydrophthalmos Norrie disease
Seizure types can include infantile spasms; generalized tonic-clonic, clonic, or tonic seizures; and myoclonic, focal, atonic, and absence seizures. Epilepsy syndromes can include Ohtahara syndrome, West syndrome, Lennox-Gaustaut syndrome, and Dravet syndrome (not SCN1A -related), classic Rett syndrome (not MECP2- related), and atypical Rett syndrome (not CDKL5 -related). ... Focal seizures were reported in about 10% of affected individuals. Epilepsy syndromes Ohtahara syndrome (OMIM 308350) is characterized by frequent generalized tonic refractory seizures and burst suppression patterns on EEG. ... Three of 80 individuals with Dravet syndrome were identified with STXBP1 encephalopathy with epilepsy [Carvill et al 2014]. Lennox-Gaustaut syndrome is characterized by multiple seizure types particularly tonic and myoclonic refractory epilepsy. ... Rett syndrome phenotype. Three individuals with the Rett syndrome phenotype have been identified with STXBP1 encephalopathy with epilepsy [Olson et al 2015, Romaniello et al 2015, Lopes et al 2016].
Early infantile epileptic encephalopathy 4 (EIEE4) is a form of early infantile epileptic encephalopathy , which refers to a group of neurological conditions characterized by severe seizures beginning in infancy. EIEE4, specifically, is often associated with partial complex or tonic-clonic seizures , although other seizure types have been reported. Other signs and symptoms may include intellectual disability, reduced muscle tone (hypotonia), hypsarrhythmia (an irregular pattern seen on EEG), dyskinesia (involuntary movement of the body), and spastic di- or quadriplegia . EIEE4 is caused by changes (mutations) in the STXBP1 gene and is inherited in an autosomal dominant manner. Treatment is based on the signs and symptoms present in each person. For example, certain medications are often prescribed to help control seizures, although they are not always effective in all people with the condition.
STXBP1 encephalopathy is a condition characterized by abnormal brain function (encephalopathy) and intellectual disability. Most affected individuals also have recurrent seizures (epilepsy). The signs and symptoms of this condition typically begin in infancy but can start later in childhood or early adulthood. In many affected individuals who have epilepsy, the seizures stop after a few years, and the other neurological problems continue throughout life. However, some people with STXBP1 encephalopathy have seizures that persist. In people with STXBP1 encephalopathy, intellectual disability is often severe to profound.
Genetic counseling. Weiss-Kruszka syndrome is inherited in an autosomal dominant manner. Approximately 95% of affected individuals have Weiss-Kruszka syndrome as the result of an apparently de novo pathogenic variant. Each child of an individual with Weiss-Kruszka syndrome has a 50% chance of inheriting the ZNF462 pathogenic variant. ... Diagnosis Formal diagnostic criteria for Weiss-Kruszka syndrome have not been established. Suggestive Findings Weiss-Kruszka syndrome should be suspected in individuals presenting with the following clinical and brain MRI findings. ... Autosomal recessive inheritance of LZTR1 -related Noonan syndrome has been reported [Johnston et al 2018].
The higher the dose used, the greater the duration of use, and the earlier age use began are predictive of worsened physical dependence and thus more severe withdrawal syndromes. Acute withdrawal syndromes can last days, weeks or months. Protracted withdrawal syndrome, also known as post-acute-withdrawal syndrome or "PAWS", is a low-grade continuation of some of the symptoms of acute withdrawal, typically in a remitting-relapsing pattern, often resulting in relapse and prolonged disability of a degree to preclude the possibility of lawful employment. Protracted withdrawal syndrome can last for months, years, or depending on individual factors, indefinitely. ... Anticonvulsants as a group however are known to cause tolerance to the anti-seizure effect. [40] SSRI drugs, which have an important use as antidepressants, engender a discontinuation syndrome that manifests with physical side effects; e.g., there have been case reports of a discontinuation syndrome with venlafaxine (Effexor). [24] See also [ edit ] Addiction Alcohol withdrawal syndrome Benzodiazepine dependence Benzodiazepine withdrawal syndrome Drug withdrawal Drug tolerance Psychological dependence Rebound insomnia Substance dependence References [ edit ] ^ Malenka RC, Nestler EJ, Hyman SE (2009). ... Retrieved 2008-12-21 . ^ Karachalios GN, Charalabopoulos A, Papalimneou V, et al. (May 2005). "Withdrawal syndrome following cessation of antihypertensive drug therapy".
It gets its name from the foot's resemblance to the bottom of a rocking chair . [1] [2] There are two subcategories of congenital vertical talus namely idiopathic or isolated type and non-idiopathic type which may be seen in association with arthrogryposis multiplex congenital, genetic syndromes and other neuromuscular disorders. [1] It can be associated with Edwards' syndrome (trisomy 18), Patau syndrome (trisomy 13), Trisomy 9 and mutation in the gene HOXD10 . [3] Contents 1 Treatment 1.1 Serial casting 1.2 Classic soft tissue release 1.3 Naviculectomy or resection arthroplasty 2 References 3 External links Treatment [ edit ] The treatment of congenital vertical talus can be broadly classified into conservative and surgical. [1] [2] [4] Serial casting [ edit ] The mainstay of management of congenital vertical talus is serial manipulative casting also known as the reversed Ponseti technique. [1] This technique involves gradual step-wise correction of the deformity usually on a weekly basis. ... S2CID 28900352 . ^ van Bosse, Harold Jacob Pieter (December 2015). "Syndromic Feet". Foot and Ankle Clinics . 20 (4): 619–644. doi : 10.1016/j.fcl.2015.07.010 . ... External links [ edit ] Classification D ICD - 9-CM : 754.61 MeSH : C536345 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum This medical sign article is a stub .
A number sign (#) is used with this entry because of evidence that isolated congenital vertical talus (CVT) is caused by heterozygous mutation in the HOXD10 gene (142984) on chromosome 2q31. Description Congenital vertical talus (CVT), also known as 'rocker-bottom foot' deformity, is a dislocation of the talonavicular joint characterized by vertical orientation of the talus with a rigid dorsal dislocation of the navicular, equinus deformity of the calcaneus, abduction deformity of the forefoot, and contracture of the soft tissues of the hind- and mid-foot. This condition is usually associated with multiple other congenital deformities and only rarely is an isolated deformity with familial occurrence (summary by Levinsohn et al., 2004). The condition is transmitted in an autosomal dominant pattern of inheritance, and sometimes shows incomplete penetrance and variable expressivity. There may be a broad spectrum of deformities, including flatfoot, talipes equinovarus (TEV or clubfoot), cavus foot, metatarsus adductus, and even hypoplasia of the tibia (summary by Dobbs et al., 2006).
Isolated congenital vertical talus (CVT) is a rare pedal deformity recognizable at birth by a dislocation of the talonavicular joint, resulting in a characteristic radiographic near-vertical orientation of the talus. Clinical description It occurs more commonly in males than females. Some patients have vertical talus in one foot and clubfoot in the other. Etiology The etiology and epidemiology of this condition are largely unknown. The reported familial cases are consistent with an autosomal dominant mode of inheritance with incomplete penetrance and mutations in HOXD10 gene have been detected in two families. Management and treatment Serial manipulation and cast immobilization followed by limited surgery provides excellent results.
It can occur by itself (isolated) or may be associated with a genetic syndrome or neuromuscular disorder. Rare familial cases have been reported, some due to a mutation in a gene called HOXD10 .
Humbert et al. (1970) first used the terms trimethylaminuria and fish-odor syndrome to describe a 6-year-old girl who intermittently had a fishy odor. She also had multiple pulmonary infections beginning in the neonatal period, the clinical stigmata of Turner syndrome but normal karyotype, splenomegaly, anemia, and neutropenia. ... Ayesh et al. (1993) studied 187 subjects with suspected body malodor ascertained in response to a newspaper story concerning the fish-odor syndrome. Biochemical tests were performed in 156 of the patients and 5 families of 6 of the subjects with the fish-odor syndrome agreed to further tests. ... All parents of 6 subjects with the syndrome who were tested showed impaired N-oxidation of excreted trimethylamine after oral challenge, indicating that they were heterozygous carriers of the allele for the syndrome. ... This demonstrated that a diagnosis of fish-odor syndrome should include the analysis of urinary excretion not only of trimethylamine but also of trimethylamine-N-oxide.
It can occur in individuals with Huntington disease or Alzheimer disease who have been given large oral therapeutic doses of choline (≥20 g/day) [Mitchell & Smith 2001, Mitchell 2005]. Olfactory reference syndrome (ORS). In some individuals who perceive that they have a body odor, clinical investigation reveals that they do not have trimethylaminuria or other body-odor disorders, but rather the psychiatric disorder ORS [McNiven et al 2019].
A rare inborn error of metabolism characterized by the presence of large amounts of trimethylamine in urine, sweat, and breath, resulting in a fishy body odor in affected individuals. While there are no additional signs and symptoms, the condition can have profound psychosocial consequences.
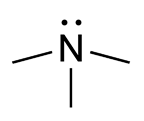
Trimethylaminuria Other names Primary trimethylaminuria Trimethylamine Specialty Endocrinology Trimethylaminuria ( TMAU ), also known as fish odor syndrome or fish malodor syndrome , [1] is a rare metabolic disorder that causes a defect in the normal production of an enzyme named flavin-containing monooxygenase 3 ( FMO3 ). [2] [3] When FMO3 is not working correctly or if not enough enzyme is produced, the body loses the ability to properly convert trimethylamine (TMA) from precursor compounds in food digestion into trimethylamine oxide (TMAO), through a process called N -oxidation . ... "Trimethylaminuria: the fish malodor syndrome". Drug Metab Dispos . 29 (4 Pt 2): 517–21. ... "Trimethylaminuria (fish malodour syndrome): a "benign" genetic condition with major psychosocial sequelae". ... Pitt JJ; Danks DM (1995). "Trimethylaminuria, fish odour syndrome: A new method of detection and response to treatment with metronidazole". ... "Trimethylaminuria: the fish-odour syndrome". Lancet . 2 (7676): 770–1. doi : 10.1016/S0140-6736(70)90241-2 .
Trimethylaminuria is a disorder in which the body is unable to break down trimethylamine, a chemical compound that has a pungent odor. Trimethylamine has been described as smelling like rotting fish, rotting eggs, garbage, or urine. As this compound builds up in the body, it causes affected people to give off a strong odor in their sweat, urine, and breath. The intensity of the odor may vary over time. The odor can interfere with many aspects of daily life, affecting a person's relationships, social life, and career. Some people with trimethylaminuria experience depression and social isolation as a result of this condition.
Trimethylaminuria causes the body to produce a fishy odor that is released in the sweat, urine, breath, and reproductive fluids. People with trimethylaminuria are unable to break down trimethylamine. Trimethylamine comes from specific chemicals (choline, carnitine, TMAO) found in certain foods. The excess trimethylamine builds up and is the source of the odor. There are no other physical symptoms from trimethylaminuria, but people with this condition may experience serious psychological and social distress. Trimethylaminuria is due to a FMO3 gene that is not working correctly.