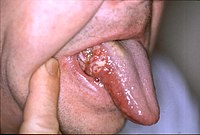
MCI can present with a variety of symptoms, and when memory loss is the predominant symptom, it is termed "amnestic MCI" and is frequently seen as a prodromal stage of Alzheimer's disease. [28] Early In people with AD, the increasing impairment of learning and memory eventually leads to a definitive diagnosis.
Overview Alzheimer's disease is a brain disorder that gets worse over time. It's characterized by changes in the brain that lead to deposits of certain proteins. Alzheimer's disease causes the brain to shrink and brain cells to eventually die. Alzheimer's disease is the most common cause of dementia — a gradual decline in memory, thinking, behavior and social skills. These changes affect a person's ability to function. About 6.5 million people in the United States age 65 and older live with Alzheimer's disease.
Alzheimer disease is a degenerative disease of the brain that causes dementia, which is a gradual loss of memory, judgment, and ability to function. This disorder usually appears in people older than age 65, but less common forms of the disease appear earlier in adulthood. Memory loss is the most common sign of Alzheimer disease. Forgetfulness may be subtle at first, but the loss of memory worsens over time until it interferes with most aspects of daily living. Even in familiar settings, a person with Alzheimer disease may get lost or become confused. Routine tasks such as preparing meals, doing laundry, and performing other household chores can be challenging.
An extreme consequence is the occurrence of the rare condition named calciphylaxis . [27] Changes in mineral and bone metabolism that may cause 1) abnormalities of calcium , phosphorus ( phosphate ), parathyroid hormone , or vitamin D metabolism; 2) abnormalities in bone turnover , mineralization , volume, linear growth, or strength ( kidney osteodystrophy ); and 3) vascular or other soft-tissue calcification. [10] CKD-mineral and bone disorders have been associated with poor outcomes. [10] Metabolic acidosis may result from decreased capacity to generate enough ammonia from the cells of the proximal tubule. [20] Acidemia affects the function of enzymes and increases excitability of cardiac and neuronal membranes by the promotion of hyperkalemia. [28] Anemia is common and is especially prevalent in those requiring haemodialysis. ... Journal of Bone and Mineral Research . 28 (1): 46–55. doi : 10.1002/jbmr.1740 .
Overview End-stage renal disease, also called end-stage kidney disease or kidney failure, occurs when chronic kidney disease — the gradual loss of kidney function — reaches an advanced state. In end-stage renal disease, your kidneys no longer work as they should to meet your body's needs. Your kidneys filter wastes and excess fluids from your blood, which are then excreted in your urine. When your kidneys lose their filtering abilities, dangerous levels of fluid, electrolytes and wastes can build up in your body. With end-stage renal disease, you need dialysis or a kidney transplant to stay alive.
Mild ventriculomegaly was seen at 21 weeks' gestation, progressive dilatation of bilateral ventricle and multiple hyperechogenic lesions at 25 weeks, and a large open cleft extending to the ependymal zone at 28 weeks. Neonatal brain CT showed bilateral open-lip schizencephaly, cortical atrophy, and absence of corpus callosum.
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( February 2018 ) Autosomal dominant porencephaly type I Other names Infantile hemiplegia with porencephaly, Porencephaly type 1 The autosomal dominant pattern of inheritance seen in this form of porencephaly [1] Autosomal dominant porencephaly type I is a rare type of porencephaly that causes cysts to grow on the brain and damage to small blood vessels, which can lead to cognitive impairment , migraines , seizures , and hemiplegia or hemiparesis . Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 4 Treatment 5 Epidemiology 6 References 7 External links Signs and symptoms [ edit ] Different people are affected very differently by this disease. The main manifestation is fluid-filled cysts that grow on the brain and can cause damage that varies depending on their location and severity. Symptoms may manifest early in infancy, or may manifest as late as adulthood. Symptoms associated with autosomal dominant porencephaly type I include migraines, hemiplegia or hemiparesis, seizures, cognitive impairment, strokes , dystonia , speech disorders , involuntary muscle spasms , visual field defects , and hydrocephalus . [2] Genetics [ edit ] Autosomal dominant porencephaly type I is caused by mutations in a gene called COL4A1 , located at 13q34 (band 34 on the long arm of chromosome 13 ).
Porencephaly Specialty Medical genetics , neurology Porencephaly is an extremely rare cephalic disorder involving encephalomalacia . [1] It is a neurological disorder of the central nervous system characterized by cysts or cavities within the cerebral hemisphere . [2] Porencephaly was termed by Heschl in 1859 to describe a cavity in the human brain. [3] Derived from Greek roots, the word porencephaly means 'holes in the brain'. [4] The cysts and cavities (cystic brain lesions) are more likely to be the result of destructive (encephaloclastic) cause, but can also be from abnormal development (malformative), direct damage, inflammation, or hemorrhage. [5] The cysts and cavities cause a wide range of physiological, physical, and neurological symptoms. [6] Depending on the patient, this disorder may cause only minor neurological problems, without any disruption of intelligence, while others may be severely disabled or face death before the second decade of their lives. However, this disorder is far more common within infants, and porencephaly can occur both before or after birth. [2] Contents 1 Signs and symptoms 2 Cause 2.1 Genetics 2.2 Cocaine and other street drugs 3 Diagnostics 4 Treatments 5 Prognosis 6 Research 7 See also 8 References 9 External links Signs and symptoms [ edit ] Patients diagnosed with porencephaly display a variety of symptoms, from mild to severe effects on the patient. Patients with severe cases of porencephaly suffer epileptic seizures and developmental delays, whereas patients with a mild case of porencephaly display little to no seizures and healthy neurodevelopment. Infants with extensive defects show symptoms of the disorder shortly after birth, and the diagnosis is usually made before the age of 1. [2] [7] The following text lists out common signs and symptoms of porencephaly in affected individuals along with a short description of certain terminologies. [2] [6] [7] [8] Degenerative or non-degenerative cavities or cysts Delayed growth and development Spastic paresis – weakness or loss in voluntary movement Contractures – painful shortening of muscles affecting motion Hypotonia – reduced muscle strength Epileptic seizures and epilepsy – multiple symptoms that involve sudden muscle spasms and loss of consciousness Macrocephaly – condition where head circumference is larger compared to other children of a certain age Microcephaly – condition where head circumference is smaller compared to other children of a certain age Hemiplegia – paralysis of appendages Tetraplegia – paralysis of limb leading to loss of function Intellectual and cognitive disability Poor or absent speech development Hydrocephalus – accumulation of cerebrospinal fluid in the brain Mental retardation Poor motor control, abnormal movements of appendages Cerebral palsy – a motor condition causing movement disabilities Blood vascular diseases such as intracerebral hemorrhage and cerebral infarction . Cerebral white-matter lesions Cause [ edit ] Porencephaly is a rare disorder.
A number sign (#) is used with this entry because of evidence that brain small vessel disease-2 (BSVD2) is caused by heterozygous mutation in the COL4A2 gene (120090) on chromosome 13q34. Description Brain small vessel disease-2 is an autosomal dominant disorder characterized by variable neurologic impairment resulting from disturbed vascular supply that leads to cerebral degeneration. The disorder is often associated with 'porencephaly' on brain imaging. Affected individuals typically have hemiplegia, seizures, and intellectual disability, although the severity is variable (summary by Yoneda et al., 2012). For a discussion of genetic heterogeneity of brain small vessel disease, see BSVD1 (175780).
In a large survey of AERD patients, it was reported that Zyflo (zileuton) was significantly more effective at controlling the symptoms of the disease than Singulair (montelukast). [28] Biologic medications such as mepolizumab (Nucala) may also be of benefit. [29] Despite optimal medical management, many patients continue to require oral steroid medications to alleviate asthma and chronic nasal congestion. [30] Surgery [ edit ] Often surgery is required to remove nasal polyps, [31] although they typically recur, particularly if aspirin desensitization is not undertaken. 90% of patients have been shown to have recurrence of nasal polyps within five years after surgery, with 47% requiring revision surgery in the same time period. [32] A complete endoscopic sinus surgery followed by aspirin desensitization has been shown to reduce the need for revision surgeries. [33] Diet [ edit ] This section needs more medical references for verification or relies too heavily on primary sources .
Uropathogenic E. coli from the gut is the cause of 80–85% of community-acquired urinary tract infections, [23] with Staphylococcus saprophyticus being the cause in 5–10%. [4] Rarely they may be due to viral or fungal infections. [24] Healthcare-associated urinary tract infections (mostly related to urinary catheterization ) involve a much broader range of pathogens including: E. coli (27%), Klebsiella (11%), Pseudomonas (11%), the fungal pathogen Candida albicans (9%), and Enterococcus (7%) among others. [6] [25] [26] Urinary tract infections due to Staphylococcus aureus typically occur secondary to blood-borne infections. [9] Chlamydia trachomatis and Mycoplasma genitalium can infect the urethra but not the bladder. [27] These infections are usually classified as a urethritis rather than urinary tract infection. [28] Intercourse In young sexually active women, sexual activity is the cause of 75–90% of bladder infections, with the risk of infection related to the frequency of sex. [4] The term "honeymoon cystitis" has been applied to this phenomenon of frequent UTIs during early marriage. ... ISBN 978-0-313-35009-2 . Archived from the original on 28 May 2016. ^ Arellano, Ronald S. (19 January 2011). ... "Prevention of urinary tract infections in persons with spinal cord injury in home health care". Home Healthcare Nurse . 28 (4): 230–41. doi : 10.1097/NHH.0b013e3181dc1bcb . ... ISBN 978-0-7817-8589-1 . Archived from the original on 28 April 2016. ^ Wilson...], [general ed.: Graham (1990).
Human papillomavirus [ edit ] Main article: HPV-positive oropharyngeal cancer Infection with human papillomavirus (HPV), particularly type 16 (there are over 180 types), is a known risk factor and independent causative factor for oral cancer. [28] A fast-growing segment of those diagnosed does not present with the historic stereotypical demographics. ... Retrieved 14 November 2017 . ^ a b "oral cancer statistics" . CancerresearchUK . Retrieved 28 October 2014 . ^ "World Life Expectancy" . 2014.
The test is recommended only when requested by a physician and should not be used to test those without symptoms. [28] Home oximetry may be effective in guiding prescription for automatically self-adjusting continuous positive airway pressure . [29] Classification [ edit ] There are three types of sleep apnea. ... Lay summary – Newswise (June 6, 2008). ^ a b c Young, Terry (28 April 2004). "Risk Factors for Obstructive Sleep Apnea in Adults". ... National Institutes of Health. 2012. Archived from the original on 28 August 2011 . Retrieved 15 February 2013 . ^ Bhimji, S. (16 December 2015). ... WebMD LLC. Archived from the original on 28 April 2016 . Retrieved 19 February 2016 . ^ Yun AJ, Lee PY, Doux JD (May 2007).
Overview Sleep apnea is a potentially serious sleep disorder in which breathing repeatedly stops and starts. If you snore loudly and feel tired even after a full night's sleep, you might have sleep apnea. The main types of sleep apnea are: Obstructive sleep apnea (OSA), which is the more common form that occurs when throat muscles relax and block the flow of air into the lungs Central sleep apnea (CSA) , which occurs when the brain doesn't send proper signals to the muscles that control breathing Treatment-emergent central sleep apnea , also known as complex sleep apnea, which happens when someone has OSA — diagnosed with a sleep study — that converts to CSA when receiving therapy for OSA If you think you might have sleep apnea, see your health care provider. Treatment can ease your symptoms and might help prevent heart problems and other complications. Symptoms The symptoms of obstructive and central sleep apneas overlap, sometimes making it difficult to determine which type you have.
Options for prophylactic treatment include the quinolone antibiotics (such as ciprofloxacin ), azithromycin , and trimethoprim/sulfamethoxazole , though the latter has proved less effective in recent years. [27] Rifaximin may also be useful. [25] [28] Quinolone antibiotics may bind to metallic cations such as bismuth, and should not be taken concurrently with bismuth subsalicylate.
Overview Traveler's diarrhea is a digestive tract disorder that commonly causes loose stools and abdominal cramps. It's caused by eating contaminated food or drinking contaminated water. Fortunately, traveler's diarrhea usually isn't serious in most people — it's just unpleasant. When you visit a place where the climate or sanitary practices are different from yours at home, you have an increased risk of developing traveler's diarrhea. To reduce your risk of traveler's diarrhea, be careful about what you eat and drink while traveling.
Relationship of AAT Protein Variants to Serum AAT Levels and Emphysema Risk in Adults View in own window AAT Protein Variant Prevalence (%) Serum AAT Levels Emphysema Risk World-wide NA Europe "True level" 1 mean (5th %ile-95th %ile) Commercial standard 2 median (5th %ile-95th %ile) MM 96.3 93.0 91.1 33 (20-53) 147 (102-254) Background MS 2.7 4.8 6.6 33 (18-52) 125 (86-218) Background MZ 0.8 2.1-3 1.9 25.4 (15-42) 90 (62-151) Background SS 0.08 0.1 0.3 28 (20-48) 95 (43-154) Background SZ 0.02 0.1 0.1 16.5 (10-23) 62 (33-108) 20%-50% ZZ 0.003 0.01 0.01 5.3 (3.4-7) ≤29 (≤29-52) 80%-100% Null-Null - - - 0 0 100% Adapted from Brantly et al [1991], Stoller & Aboussouan [2005], de Serres & Blanco [2012], Bornhorst et al [2013] AAT = alpha-1 antitrypsin; NA = North America 1. µmol/L 2. mg/dL Note: An attempt to correlate serum AAT levels with protein variants in children showed trends similar to those seen in adults [Donato et al 2012].
Alpha-1 antitrypsin deficiency (AATD) is an inherited disease that causes an increased risk of having chronic obstructive pulmonary disease (COPD), liver disease , skin problems ( panniculitis ), and inflammation of the blood vessels ( vasculitis ). Lung (pulmonary) problems almost always occur in adults, whereas liver and skin problems may occur in adults and children. The age symptoms begin and severity of symptoms can vary depending on how much working alpha-1 antitrypsin protein (AAT) a person has. Symptoms may include shortness of breath and wheezing, repeated infections of the lungs and liver, yellow skin, feeling overly tired ( fatigue ), rapid heartbeat when standing, vision problems, and weight loss. However, some people with AATD do not have any problems. AATD is caused by changes (pathogenic variants, also called mutations) in the SERPINA1 gene and it is inherited in a codominant manner.
A number sign (#) is used with this entry because alpha-1-antitrypsin deficiency is caused by mutation in the SERPINA1 gene (107400), most often by homozygosity for the PiZ allele (107400.0011). Description Alpha-1-antitrypsin deficiency is an autosomal recessive disorder. The most common manifestation is emphysema, which becomes evident by the third to fourth decade. A less common manifestation of the deficiency is liver disease, which occurs in children and adults, and may result in cirrhosis and liver failure. Environmental factors, particularly cigarette smoking, greatly increase the risk of emphysema at an earlier age (Crystal, 1990).
A hereditary disease that develops in adulthood and is characterized by chronic liver disorders (cirrhosis), respiratory disorders (emphysema), and rarely panniculitis.
Alpha-1 antitrypsin deficiency is an inherited disorder that may cause lung disease and liver disease. The signs and symptoms of the condition and the age at which they appear vary among individuals. People with alpha-1 antitrypsin deficiency usually develop the first signs and symptoms of lung disease between ages 20 and 50. The earliest symptoms are shortness of breath following mild activity, reduced ability to exercise, and wheezing. Other signs and symptoms can include unintentional weight loss, recurring respiratory infections, fatigue, and rapid heartbeat upon standing.
Best known for portraying Harry Bentley, The Jeffersons English next door neighbour [27] Big Show (born Paul Wight; 1972), American professional wrestler and actor, currently working for the WWE , had his pituitary tumor removed in 1991. [28] Eddie Carmel , born Oded Ha-Carmeili (1936–1972), Israeli-born entertainer with gigantism and acromegaly, popularly known as "The Jewish Giant" Ted Cassidy (1932–1979), American actor.
Overview Acromegaly is a hormonal disorder that develops when your pituitary gland produces too much growth hormone during adulthood. When you have too much growth hormone, your bones increase in size. In childhood, this leads to increased height and is called gigantism. But in adulthood, a change in height doesn't occur. Instead, the increase in bone size is limited to the bones of your hands, feet and face, and is called acromegaly. Because acromegaly is uncommon and the physical changes occur slowly over many years, the condition sometimes takes a long time to recognize. Untreated, high levels of growth hormone can affect other parts of the body, in addition to your bones.
A number sign (#) is used with this entry because of evidence that growth hormone-secreting pituitary adenoma-2 (PITA2) is caused by mutation in the GPR101 gene (300393) on chromosome Xq26. For a general phenotypic description and a discussion of genetic heterogeneity of pituitary adenoma, see 102200. Molecular Genetics Because GPR101 was implicated as the gene that drives the phenotype of X-linked early-onset gigantism related to a microduplication (see 300942), Trivellin et al. (2014) sequenced the GPR101 gene in 248 sporadic acromegaly patients. Trivellin et al. (2014) identified a recurrent GPR101 mutation (E308D; 300393.0001) in 11 of 248 sporadic acromegaly patients with somatic somatotropinoma. Three of these 11 appeared to carry a constitutive mutation that was detected in DNA from peripheral blood mononuclear cells (PBMCs).
A rare acquired endocrine disease related to excessive production of growth hormone (GH) and characterized by progressive somatic disfigurement (mainly involving the face and extremities) and systemic manifestations. Epidemiology Worldwide, the prevalence is 1/7,500 to 1/35,800. The annual incidence is 1/91,000 to 1/526,000. Clinical description Due to its insidious onset and slow progression, acromegaly is often diagnosed from four to more than ten years after its onset, and is most often diagnosed in middle age (average age 40-50 years). The main clinical features are broadened extremities (hands and feet), widened, thickened and stubby fingers, and thickened soft tissue. The facial aspect is characteristic and includes a widened and thickened nose, prominent cheekbones, forehead bulges, thick lips and marked facial lines.
Acromegaly occurs when the pituitary gland makes too much growth hormone (GH). It is most often diagnosed in middle-aged adults, although symptoms can appear at any age. Signs and symptoms include abnormal growth of the hands and feet and overgrowth of the bones in the face. Other symptoms may include joint pain, headache, backbone fractures, high blood pressure, and diabetes. Acromegaly is most often caused by non-cancerous tumors on the pituitary called adenomas.
Because most of the mutation-positive tumors were adenocarcinomas from 'never smokers' (defined as patients who smoked less than 100 cigarettes in a lifetime), Pao et al. (2004) screened EGFR exons 2 through 28 for mutations in 15 adenocarcinomas resected from untreated 'never smokers.'
Lung cancer is a disease in which certain cells in the lungs become abnormal and multiply uncontrollably to form a tumor . Lung cancer may not cause signs or symptoms in its early stages. Some people with lung cancer have chest pain, frequent coughing, blood in the mucus, breathing problems, trouble swallowing or speaking, loss of appetite and weight loss, fatigue, or swelling in the face or neck. Additional symptoms can develop if the cancer spreads (metastasizes ) into other tissues. Lung cancer occurs most often in adults in their sixties or seventies. Most people who develop lung cancer have a history of long-term tobacco smoking; however, the condition can occur in people who have never smoked.
Risk factors There are many risk factors for heart diseases: age, sex, tobacco use, physical inactivity, excessive alcohol consumption, unhealthy diet, obesity, genetic predisposition and family history of cardiovascular disease, raised blood pressure ( hypertension ), raised blood sugar ( diabetes mellitus ), raised blood cholesterol ( hyperlipidemia ), undiagnosed celiac disease , psychosocial factors, poverty and low educational status, and air pollution . [14] [15] [16] [17] [18] While the individual contribution of each risk factor varies between different communities or ethnic groups the overall contribution of these risk factors is very consistent. [19] Some of these risk factors, such as age, sex or family history/genetic predisposition, are immutable; however, many important cardiovascular risk factors are modifiable by lifestyle change, social change, drug treatment (for example prevention of hypertension, hyperlipidemia, and diabetes). [20] People with obesity are at increased risk of atherosclerosis of the coronary arteries . [21] Genetics Genetic factors influence the development of cardiovascular disease in men who are less than 55 years old and in women who are less than 65 years old. [20] Cardiovascular disease in a person's parents increases their risk by 3 fold. [22] Multiple single nucleotide polymorphisms (SNP) have been found to be associated with cardiovascular disease in genetic association studies, [23] [24] but usually, their individual influence is small, and genetic contributions to cardiovascular disease are poorly understood. [24] Age Calcified heart of an older woman with cardiomegaly Age is the most important risk factor in developing cardiovascular or heart diseases, with approximately a tripling of risk with each decade of life. [25] Coronary fatty streaks can begin to form in adolescence. [26] It is estimated that 82 percent of people who die of coronary heart disease are 65 and older. [27] Simultaneously, the risk of stroke doubles every decade after age 55. [28] Multiple explanations are proposed to explain why age increases the risk of cardiovascular/heart diseases.
However, the introduction of combination therapy with concurrent rituximab and cladribine therapy has shown excellent results in early follow-up. [28] As of 2016, this therapy is considered the first-line treatment of choice for many people with HCL-V. [29] Many older treatment approaches, such as Interferon - alpha , the combination chemotherapy regimen "CHOP", and common alkylating agents like cyclophosphamide showed very little benefit. [26] Pentostatin and cladribine administered as monotherapy (without concurrent rituximab) provide some benefit to many people with HCL-V, but typically induce shorter remission periods and lower response rates than when they are used in classic HCL.
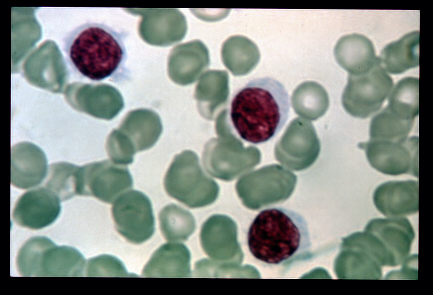
Hairy cell leukemia is a rare, slow-growing cancer of the blood in which the bone marrow makes too many B cells (lymphocytes), a type of white blood cell that fights infection. The condition is named after these excess B cells which look 'hairy' under a microscope. As the number of leukemia cells increases, fewer healthy white blood cells, red blood cells and platelets are produced. The symptoms include a large spleen (splenomegalia) but without an increase of lymph nodes, and general symptoms such as fever, night sweats, fatigue, weight loss. Blood exams show the decreased number of blood cells and platelets. The diagnosis can be made with the biopsy of the bone marrow, blood exams showing antigens that are released by the B-cells.
A rare, slowly progressive, chronic leukemia characterized by presence of abnormal B-lymphocytes (medium sized with abundant irregular pale cytoplasm, hair-like cytoplasmic projections/ruffled cytoplasmic border, a round or bean-shaped nucleus and absent nucleoli) in the blood or bone marrow, spleen and peripheral blood pancytopenia, notable monocytopenia, and marked susceptibility to infection. The characteristic immunophenotype is CD11c+, CD25+, CD103+ and CD123+ with a BRAF mutation in most cases. Epidemiology Classic hairy cell leukemia (HCLc) accounts for 2% of all leukemia cases. Estimates of the annual incidence range between 1/213,000-2,860,000 worldwide. HCLc is observed more commonly in caucasians. Men are predominantly affected with a male:female ratio of 4:1.
Overview Hairy cell leukemia is a cancer of the white blood cells. The white blood cells help fight off germs. There are a few different types of white blood cells. The white blood cells involved in hairy cell leukemia are called B cells. B cells are also called B lymphocytes. In hairy cell leukemia, the body makes too many B cells . The cells don't look like healthy B cells . Instead, they've undergone changes to become leukemia cells. The leukemia cells look "hairy" under a microscope. Hairy cell leukemia cells keep living when healthy cells would die as part of the natural cell life cycle.
However, the exact roles, if any, of these genomic abnormalities in promoting the progression of ISFL to FL are unclear. [24] Transformed follicular lymphoma [ edit ] The transformation of FL to a more aggressive state or other type of aggressive lymphoma is associated with: 1) primarily gene-activating mutations in CREEBP, KMT2D, STAT6, CARD11 (encoding a guanylate kinase which interacts with BCL10 and activates NF-κB to regulate cell survival); 2) changes in the expression of diverse genes; 3) the overproduction of various cell-activating cytokines [25] and CD79B (encoding the Ig-beta protein component of the B-cell receptor [26] ); 4) gene-inactivating mutations in TNFAIP3, CD58 (encoding the cell adhesion molecule , lymphocyte function-associated antigen 3, that is involved in activating T-cells [27] ), CDKN2A (encoding p16INK4a and p14arf tumor suppressor proteins [28] ) or CDKN2B (encoding cyclin dependent kinase inhibitor 2B multiple tumor suppressor 2 [29] ) (inactivation of either CDKN2 gene causes genome instability , i.e. increased frequency of other gene mutations), and TNFRSF4 (encoding one type of tumor necrosis factor receptor [30] ); and 5) gene-activating or -inactivating mutations in, or other causes for the under- or over-expression of, c-MYC ((encoding the c-Myc proto- oncogene transcription factor that regulates the expression of diverse genes many of which promote cell proliferation [31] ). [24] Tumor environment [ edit ] The non-neoplastic immune and stromal cells as well as the extracellular matrix in tissues may enable neoplastic follicular cells to survive, proliferate, and avoid surveillance by the immune system . ... Phosphoionsitide 3-kinase inhibitors produced overall response rates of 10–12.5 months in 42-59%; tisagenlecleuce cells produced an overall progression-free response rate of 70% after a follow-up of 28 months; [52] phosphoinositide 3-kinase inhibitors produced overall response rates of ~40% and complete response rates of 1-20%; Bruton's tyrosine kinase inhibitor produced overall and complete response rates of 38% and 18%, respectively; the Bcl inhibitor produce overall and complete response rates of 33% and 14%, respectively; histone deacetylase inhibitors produce overall response rates of 35%-71%; and checkpoint inhibitors produce overall response rates of 40%-80% and complete response rates of 10-60%. [4] See also [ edit ] List of hematologic conditions large-cell lymphoma In situ follicular lymphoma References [ edit ] ^ a b c d e f g h Xerri L, Dirnhofer S, Quintanilla-Martinez L, Sander B, Chan JK, Campo E, et al.
Follicular lymphoma is a form of non-Hodgkin lymphoma (see this term) characterized by a proliferation of B cells whose nodular structure of follicular architecture is preserved. Epidemiology Prevalence of follicular lymphoma is estimated at about 1/3,000. Clinical description The median age at diagnosis is 60-65 years. The disease is extremely rare in children. Follicular lymphoma is located primarily in the lymph nodes, but can also involve the spleen, bone marrow, peripheral blood and Waldeyer's ring. The skin and central nervous system are affected in rare cases. Symptoms appear at an advanced stage of the disease and can include fever, night sweats and weight loss.
Description Follicular non-Hodgkin lymphoma is an indolent B-cell malignancy with an annual incidence exceeding 10,000 cases in the United States (Bohen et al., 2003). One form of susceptibility to follicular lymphoma (FL1) is associated with a region on chromosome 6p21.33. Mapping In a subtype-specific genomewide association study of non-Hodgkin lymphoma susceptibility, Skibola et al. (2009) found that the single-nucleotide polymorphism (SNP) rs6457327 on chromosome 6p21.33 was associated with susceptibility to follicular lymphoma in 189 cases and 592 controls, with validation in another 456 follicular lymphoma cases and 2,785 controls (combined allelic P = 4.7 x 10(-11)). The SNP rs6457327 is 5 kb downstream of the 3-prime untranslated region of the C6ORF15 gene (STG; 611401) and telomeric to HLA-C (see 142840) in the major histocompatibility complex, an allele of which influences susceptibility to psoriasis (see PSORS1, 177900). In a 3-stage genomewide association study to identify susceptibility loci for non-Hodgkin lymphoma (NHL) subtypes, Conde et al. (2010) identified 2 variants associated with follicular lymphoma at 6p21.32, rs10484561 (combined p = 1.12 x 10(-29)) and rs7755224 (combined p = 2.00 x 10(-19)), supporting the idea that major histocompatibility complex (MHC) genetic variation influences follicular lymphoma susceptibility.
Furthermore, it secretes TIMP 1 and 2, naturally occurring inhibitors of matrix metalloproteinases , which prevents them from breaking down the fibrotic material in the extracellular matrix . [27] [28] As this cascade of processes continues, fibrous tissue bands (septa) separate hepatocyte nodules, which eventually replace the entire liver architecture, leading to decreased blood flow throughout. ... Archived from the original on 2008-05-28. ^ Muriel, P; Arauz, J (Jul 2010).
Overview Cirrhosis is severe scarring of the liver. This serious condition can be caused by many forms of liver diseases and conditions, such as hepatitis or chronic alcoholism. Each time your liver is injured — whether by excessive alcohol consumption or another cause, such as infection — it tries to repair itself. In the process, scar tissue forms. As cirrhosis gets worse, more and more scar tissue forms, making it difficult for the liver to do its job. Advanced cirrhosis is life-threatening. The liver damage caused by cirrhosis generally can't be undone. But if liver cirrhosis is diagnosed early and the underlying cause is treated, further damage can be limited.
Conducted by the agency's Committee for Medicinal Products for Human Use (CHMP), the review encompassed available data from the companies that market these drugs, postmarketing safety data, randomized controlled studies, 2 studies of unlicensed oral calcitonin drugs, and experimental cancer studies, among other sources. [ citation needed ] In 2014, the FDA noted the risk imbalances in the prescribing information for Miacalcin but declined to label this product with a boxed warning as a causal association was not identified. [26] A more recent meta-analysis determined that a causal link between calcitonin and cancer is both unlikely and antithetical to known biology, although a weak association was not definitively excluded. [27] The available studies for analysis were inconsistent and nonspecific, with one study [28] noting an increased risk of liver cancer and decreased risk of breast cancer.
Overview Paget's (PAJ-its) disease of bone interferes with your body's normal recycling process, in which new bone tissue gradually replaces old bone tissue. Over time, bones can become fragile and misshapen. The pelvis, skull, spine and legs are most commonly affected. The risk of Paget's disease of bone increases with age and if family members have the disorder. However, for reasons unknown to doctors, the disease has become less common over the past several years and is less severe when it does develop. Complications can include broken bones, hearing loss and pinched nerves in your spine.
None have currently been widely adopted as a complete model. [28] Signs and symptoms A man with Parkinson's disease displaying a flexed walking posture pictured in 1892 [29] Handwriting of a person affected by PD [30] Main article: Signs and symptoms of Parkinson's disease The most recognizable symptoms in Parkinson's disease are movement ("motor") related. [31] Non-motor symptoms, which include autonomic dysfunction, neuropsychiatric problems (mood, cognition, behavior or thought alterations), and sensory (especially altered sense of smell) and sleep difficulties, are also common.
Overview Parkinson's disease is a progressive disorder that affects the nervous system and the parts of the body controlled by the nerves. Symptoms start slowly. The first symptom may be a barely noticeable tremor in just one hand. Tremors are common, but the disorder may also cause stiffness or slowing of movement. In the early stages of Parkinson's disease, your face may show little or no expression. Your arms may not swing when you walk. Your speech may become soft or slurred.
Of the relatives of 29 PCO probands, 15 of 29 (52%) mothers, 6 of 28 (21%) fathers, 35 of 53 (66%) sisters, and 4 of 18 (22%) brothers were assigned affected status.
Overview Polycystic ovary syndrome (PCOS) is a problem with hormones that happens during the reproductive years. If you have PCOS , you may not have periods very often. Or you may have periods that last many days. You may also have too much of a hormone called androgen in your body. With PCOS , many small sacs of fluid develop along the outer edge of the ovary. These are called cysts. The small fluid-filled cysts contain immature eggs.
Polycystic ovary syndrome is a condition that affects women in their child-bearing years and alters the levels of multiple hormones, resulting in problems affecting many body systems. Most women with polycystic ovary syndrome produce excess male sex hormones (androgens), a condition called hyperandrogenism. Having too much of these hormones typically leads to excessive body hair growth (hirsutism), acne, and male pattern baldness. Hyperandrogenism and abnormal levels of other sex hormones prevent normal release of egg cells from the ovaries (ovulation) and regular menstrual periods, leading to difficulty conceiving a child (subfertility) or a complete inability to conceive (infertility). For those who achieve pregnancy, there is an increased risk of complications and pregnancy loss.
It is the most common cause of delayed puberty in girls [1] [8] (30%) [7] and even more so in boys [2] (65%). [10] It is commonly inherited with as much as 80% of the variation in the age of onset of puberty due to genetic factors. [10] [12] These children have a history of shorter stature than their age-matched peers throughout childhood, but their height is appropriate for bone age , meaning that they have delayed skeletal maturation with potential for future growth. [7] It is often difficult to establish if it is a true constitutional delay of growth and puberty or if there is an underlying pathology because lab tests are not always discriminatory. [13] In absence of any other symptoms, short stature, delayed growth in height and weight, and/or delayed puberty may be the only clinical manifestations of certain chronic diseases including coeliac disease . [14] [15] [16] [17] Malnutrition or chronic disease [ edit ] When underweight or sickly children present with pubertal delay, it is warranted to search for illnesses that cause a temporary and reversible delay in puberty. [2] Chronic conditions such as sickle cell disease [18] [19] [20] and thalassemia , [21] cystic fibrosis , [22] HIV/AIDS , hypothyroidism , [23] chronic kidney disease , [24] [25] and chronic gastroenteric disorders (such as coeliac disease [15] [26] and inflammatory bowel disease [27] [28] [29] ) cause a delayed activation of the hypothalamic region of the brain to send signals to start puberty. [30] Childhood cancer survivors can also present with delayed puberty secondary to their cancer treatments, especially males. [10] [31] The type of treatment, amount of exposure/dosage of drugs, and age during treatment determine the level by which the gonads are affected with younger patients at a lower risk of negative reproductive effects. [31] Excessive physical exercise and physical stress, especially in athletes can also delay pubertal onset. [32] Eating disorders such as bulimia nervosa and anorexia nervosa can also impair puberty due to undernutrition . [30] [33] Carbohydrate-restricted diets for weight loss has also been shown to decrease the stimulation of insulin which in turn does not stimulate kisspeptin neurons vital in the release of puberty-starting hormones. [34] This shows that carbohydrate restricted children and children with diabetes mellitus type 1 can have delayed puberty. [11] [35] Primary failure of the ovaries or testes (hypergonadotropic hypogonadism) [ edit ] Hypothalamic-pituitary-testicular axis and the hormones produced by each part of the axis.
Accurate diagnosis is usually accomplished through immunohistochemical staining for muscle-specific proteins such as myogenin , muscle-specific actin , desmin , D-myosin , and myoD1 . [18] [27] [28] Myogenin, in particular, has been shown to be highly specific to RMS, [29] although the diagnostic significance of each protein marker may vary depending on the type and location of the malignant cells.
A number sign (#) is used with this entry because of evidence that rhabdomyosarcoma can be caused by somatic mutation in the SLC22A18 gene (602631) on chromosome 11p15. Mapping Scrable et al. (1987) generalized the hypothesis of somatic chromosomal interchanges at mitosis as the basis of retinoblastoma (180200). To the study of other tumors, the proposed model suggests that predisposing recessive mutations are revealed by exchanges that result in homozygous (or hemizygous) defect at the tumor locus. This would suggest that for tumors with no known cytogenetic aberrations, one could find the location of these genes by delineating the smallest overlapping region of genetic homozygosity shared among tumors of the same type. They applied this rationale to rhabdomyosarcoma, a 'pediatric tumor.'
A malignant soft tissue tumor which develops from cells of striated muscle. It is the most common form of tumor found in children and adolescents. Epidemiology The annual incidence is 1/170,000. In children younger than 15 years, the annual incidence is estimated at 1/244,000. Clinical description The median age of diagnosis is 5 years. Rhabdomyosarcoma can develop anywhere in the body, including in sites where striated muscle does not normally occur. The most frequent locations are: the head and neck (40%), including tumors of the orbit and para-meningeal tumors, the genitourinary tract (20%) including tumors in the bladder and/or prostate, in the uterus and vagina, para-testicular tumors, and tumors in the limbs (20%) and trunk (10%).
Overview Rhabdomyosarcoma (RMS) is a rare type of cancer that forms in soft tissue — specifically skeletal muscle tissue or sometimes hollow organs such as the bladder or uterus. RMS can occur at any age, but it most often affects children. Although RMS can arise anywhere in the body, it's more likely to start in the: Head and neck area Urinary system, such as the bladder Reproductive system, such as the vagina, uterus and testes Arms and legs The outlook (prognosis) and treatment decisions depend on the type of rhabdomyosarcoma, where it starts, tumor size and whether the cancer has spread. Treatment is usually with a combination of surgery, chemotherapy and radiation therapy. Major advancements in the treatment of rhabdomyosarcoma have significantly improved outcomes. After completion of treatment, people need lifelong monitoring for potential late effects of intense chemotherapy and radiation.