Acute motor axonal neuropathy Other names Acute pure motor Guillain-Barré syndrome Specialty Neurology Acute motor axonal neuropathy (AMAN) is a variant of Guillain–Barré syndrome . ... Several of the paralysis patients were found to have antibodies to C. jejuni and anti-GD1a antibodies, suggesting a link between the pathogen and the disease. [1] In 2015, Zika virus was linked to AMAN. [2] Diagnosis [ edit ] The syndrome typically presents as a progressive flaccid symmetric paralysis with areflexia , often causing respiratory failure. ... You can help by adding to it . ( October 2017 ) History [ edit ] AMAN, also known as Chinese Paralytic Syndrome , [3] was first described by a group of Johns Hopkins University and University of Pennsylvania neurologists in collaboration with neurologists from the Second Teaching Hospital of Hebei Medical School and Beijing Children's Hospital . [4] In 1991, Guy Mckhann, Jack Griffin, Dave Cornblath and Tony Ho from Johns Hopkins University and Arthur Asbury from University of Pennsylvania visited China to study a mysterious epidemic of paralytic syndrome occurring in northern China. ... In contrast, the common form of Guillain–Barré syndrome in the West often presents with sensory loss and demyelination on electrophysiology testing and is more common in adults. ... PMID 1679153 . ^ Griffin JW, Li CY, Ho TW, Xue P, Macko C, Gao CY, Yang C, Tian M, Mishu B, Cornblath DR, et al. (1995). "Guillain–Barré syndrome in northern China: The spectrum of neuropathological changes in clinically defined cases".
A rare pure motor axonal form of Guillain-Barré syndrome (GBS). Epidemiology The overall annual incidence of GBS is estimated at between 1/91,000 and 1/55,000.
CHAPLE Syndrome Specialty Medical genetics Symptoms Gastrointestinal symptoms, edema, malnutrition, hypoalbuminemia, hypogammaglobulinemia, intestinal lymphangiectasia [1,2] Duration Lifelong Causes Genetic (autosomal recessive) Diagnostic method Genetic testing Treatment Eculizumab CD55 deficiency , also called DAF deficiency or CHAPLE syndrome , is a rare genetic disorder of the immune system . ... Symptoms can include abdominal pain , nausea , vomiting , diarrhea , loss of appetite , weight loss , and edema . [1] People also have chronic malabsorption , which causes deficiencies in iron , ferritin , calcium , magnesium , folate , vitamin D and vitamin B12 . [1] Some patients may have recurrent respiratory infections associated with hypogammaglobulinemia . [1] Severe thrombotic vascular occlusions may also be found among these patients. [1] Genetics [ edit ] CHAPLE Syndrome has an autosomal recessive pattern of inheritance CHAPLE syndrome is caused by mutations of the complement regulator CD55 gene leading to a loss of protein expression. [1] Inheritance [ edit ] CHAPLE syndrome is primarily inherited in an autosomal recessive manner. [1] This means that usually a child inherits a copy of the mutated gene from both parents, resulting in a homozygous defect. [2] Pathophysiology [ edit ] CHAPLE syndrome is characterized by complement-mediated autoimmune hemolysis and paroxysmal nocturnal hemoglobinuria . ... When CD55 is absent, the complement system attacks red blood cells and causes them to be destroyed (hemolysis). [3] [4] [5] Diagnosis [ edit ] CHAPLE syndrome patients are generally diagnosed through a combination of clinical presentation, histology, and genetic testing. ... PMID 26582375 . ^ Ozen A (January 2019). "CHAPLE syndrome uncovers the primary role of complement in a familial form of Waldmann's disease".
A number sign (#) is used with this entry because of evidence that Ververi-Brady syndrome (VERBRAS) is caused by heterozygous mutation in the QRICH1 gene (617387) on chromosome 3p21. Description Ververi-Brady syndrome is characterized by mild developmental delay, mildly impaired intellectual development and speech delay, and mild dysmorphic facial features. ... Lui et al. (2019) identified 2 unrelated children, an 8-year-old girl and a 11 year-old boy, with features of Ververi-Brady syndrome, including short stature, irregular growth plates of the proximal phalanges, developmental delay, and mild dysmorphic facial features. ... Molecular Genetics In 3 unrelated patients with Ververi-Brady syndrome (VERBRAS; 617982), Ververi et al. (2018) identified de novo heterozygous nonsense or frameshift mutations in the QRICH1 gene (617387.0001-617387.0003). ... Lui et al. (2019) also identified an 11-year-old boy in the DECIPHER database (Firth et al., 2009) with features of VERBRAS syndrome and a de novo nonsense mutation in the QRICH1 gene (R511X).
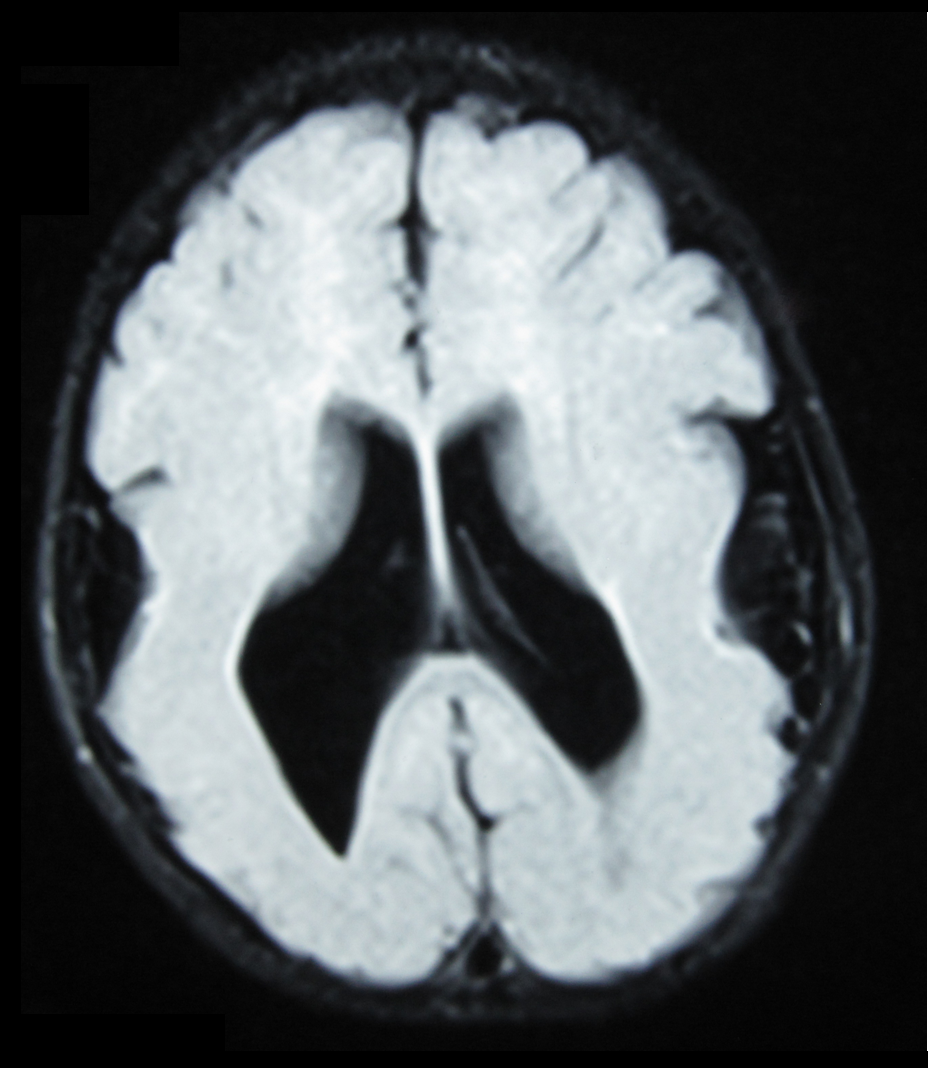
Contents 1 Symptoms 2 Cause 3 Diagnosis 3.1 Types of NMD syndromes 4 Treatment 5 Prognosis 6 References Symptoms [ edit ] Symptoms vary according to the abnormality, but often feature poor muscle tone and motor function, seizures , developmental delays , mental retardation , failure to grow and thrive, difficulties with feeding, swelling in the extremities, and a smaller than normal head. ... Abnormal migration ultimately results in abnormal gyral formation. [4] Diagnosis [ edit ] Types of NMD syndromes [ edit ] More than 25 syndromes resulting from abnormal neuronal migration have been described. Among them are syndromes with several different patterns of inheritance; genetic counseling thus differs greatly between syndromes. [2] Lissencephaly Microlissencephaly Schizencephaly Porencephaly Pachygyria Polymicrogyria Agyria Macrogyria Microgyria Micropolygyria Grey matter heterotopia Agenesis of the corpus callosum Agenesis of the cranial nerves Band heterotopias Focal cortical dysplasia , Miller–Dieker syndrome , muscle-brain-eye syndrome [ de ] , Fukuyama congenital muscular dystrophy and Walker–Warburg syndrome are genetic disorders associated with lissencephaly. [5] Treatment [ edit ] Treatment is symptomatic and may include anti-seizure medication and special or supplemental education consisting of physical , occupational and speech therapies . [2] Prognosis [ edit ] The prognosis for children with NMDs varies depending on the specific disorder and the degree of brain abnormality and subsequent neurological signs and symptoms. [2] References [ edit ] ^ Sarnat, Harvey (1992).
Find sources: "Bhaskar–Jagannathan syndrome" – news · newspapers · books · scholar · JSTOR ( June 2012 ) ( Learn how and when to remove this template message ) This article needs attention from an expert on the subject . ... Please help to improve this article by introducing more precise citations. ( July 2018 ) ( Learn how and when to remove this template message ) ( Learn how and when to remove this template message ) Bhaskar–Jagannathan syndrome Specialty Genetic disorder Bhaskar–Jagannathan syndrome is an extremely rare genetic disorder and there is a limited amount of information related to it. ... Treatment [ edit ] Treatment for this rare genetic disorder can be physical therapy , there have been antibiotics found to be effective, and surgery has been found to be another solution. [1] [2] References [ edit ] ^ Bhaskar Jagannathan Syndrome (2012). In IAMUNWELL. Retrieved April 5, 2012, from http://www.iamunwell.com/Diseases-Alphabet-B/bhaskar-jagannathan-syndrome.html Archived 2012-08-13 at the Wayback Machine ^ Bhaskar Jagannathan syndrome .
A number sign (#) is used with this entry because of evidence that Kleefstra syndrome-2 (KLEFS2) is caused by heterozygous mutation in the KMT2C gene (606833) on chromosome 7q36. Description Kleefstra syndrome-2 is an autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, variable intellectual disability, and mild dysmorphic features (summary by Koemans et al., 2017). For a discussion of genetic heterogeneity of Kleefstra syndrome, see KLEFS1 (610253). Clinical Features Kleefstra et al. (2012) reported a girl with Kleefstra syndrome-2. ... Molecular Genetics In 4 of 9 EHMT1 (607001) mutation-negative patients with core features of Kleefstra syndrome-1 (KLEFS1; 610253) but otherwise heterogeneous phenotypes, Kleefstra et al. (2012) identified mutations in 4 functionally related genes, KMT2C (606833.0001), MBD5 (611472), SMARCB1 (601607), and NR1I3 (603881).
A number sign (#) is used with this entry because of evidence that presynaptic congenital myasthenic syndrome-7 (CMS7) is caused by heterozygous mutation in the SYT2 gene (600104) on chromosome 1q32. Description Congenital myasthenic syndromes (CMS) are a group of inherited disorders affecting the neuromuscular junction (NMJ). ... Clinical Features Herrmann et al. (2014) reported 2 unrelated multigenerational families with congenital myasthenic syndrome. The first family was from the United States and contained 4 patients who presented in early childhood with foot deformities, including pes cavus and hammertoes. ... Repetitive nerve stimulation resulted in large decrements of the motor amplitude, consistent with myasthenic syndrome, and maximum voluntary contraction resulted in posttetanic potentiation lasting up to 60 minutes. ... Inheritance The transmission pattern of presynaptic congenital myasthenic syndrome in the families reported by Herrmann et al. (2014) was consistent with autosomal dominant inheritance.
A number sign (#) is used with this entry because of evidence that congenital myasthenic syndrome-23 (CMS23) is caused by homozygous mutation in the SLC25A1 gene (190315) on chromosome 22q11. ... Chaouch et al. (2014) found that another unrelated 2-year-old patient with D2L2AD (Edvardson et al., 2013) had markedly increased jitter and blocking on EMG, consistent with myasthenic syndrome. Her symptoms did not fluctuate and did not improve with 3,4-DAP and ephedrine. ... INHERITANCE - Autosomal recessive HEAD & NECK Eyes - Ptosis Neck - Neck muscle weakness SKELETAL Feet - Foot deformities MUSCLE, SOFT TISSUES - Myasthenic syndrome - Muscle weakness - Easy fatigability - Hypotonia, distal and proximal - Abnormal jitter seen on EMG - Calf hypertrophy NEUROLOGIC Central Nervous System - Delayed motor development - Delayed walking - Frequent falls - Impaired intellectual development, mild - Learning disabilities Peripheral Nervous System - Hyporeflexia MISCELLANEOUS - Onset in early infancy - Nonprogressive disease course - Favorable response to treatment with 3,4-DAP or pyridostigmine - Two sibs born of consanguineous parents have been reported (last curated November 2018) MOLECULAR BASIS - Caused by mutation in the solute carrier family 25 (mitochondrial carrier, citrate transporter), member 1 gene (SLC25A1, 190315.0007 ) ▲ Close
A number sign (#) is used with this entry because of evidence that congenital myasthenic syndrome-18 (CMS18) is caused by heterozygous mutation in the SNAP25 gene (600322) on chromosome 20p11. ... Description Congenital myasthenic syndrome-18 is an autosomal dominant presynaptic neuromuscular disorder characterized by early-onset muscle weakness and easy fatigability associated with delayed psychomotor development and ataxia (summary by Shen et al., 2014).
A number sign (#) is used with this entry because of evidence that congenital myasthenic syndrome-8 (CMS8) is caused by homozygous or compound heterozygous mutation in the AGRN gene (103320) on chromosome 1p36. Description Congenital myasthenic syndromes are genetic disorders of the neuromuscular junction (NMJ) that are classified by the site of the transmission defect: presynaptic, synaptic, and postsynaptic. ... Clinical Features Huze et al. (2009) reported a brother and sister, born of distantly related Swiss parents, with congenital myasthenic syndrome. The proband was a 42-year-old woman who reported inability to run and ptosis from early childhood. ... Inheritance The transmission pattern of congenital myasthenic syndrome in the family reported by Huze et al. (2009) was consistent with autosomal recessive inheritance. ... In a man with congenital myasthenic syndrome, Maselli et al. (2012) identified compound heterozygosity for a missense and a nonsense mutation in the AGRN gene (V1727F, 103320.0002 and Q353X, 103320.0003).
A number sign (#) is used with this entry because of evidence that congenital myasthenic syndrome-19 (CMS19) is caused by homozygous mutation in the COL13A1 gene (120350) on chromosome 10q22. Description Congenital myasthenic syndrome-19 is an autosomal recessive disorder resulting from a defect in the neuromuscular junction, causing generalized muscle weakness, exercise intolerance, and respiratory insufficiency. ... Clinical Features Logan et al. (2015) reported 3 patients from 2 unrelated families with onset of congenital myasthenic syndrome soon after birth. Two unrelated patients were aged 2 years and 27 years at the time of the report; the younger sib of the older patient had died of respiratory insufficiency at age 8 years. ... Repetitive nerve stimulation performed in 2 unrelated patients showed a significant decremental response, consistent with a congenital myasthenic syndrome. Clinical Management One of the patients with CMS19 reported by Logan et al. (2015) had motor and respiratory improvement with 3,4-diaminopyridine and salbutamol treatment.
A number sign (#) is used with this entry because presynaptic congenital myasthenic syndrome-6 (CMS6) is caused by homozygous or compound heterozygous mutation in the choline acetyltransferase gene (CHAT; 118490) on chromosome 10q11. Description Congenital myasthenic syndromes (CMS) are a group of inherited disorders affecting the neuromuscular junction (NMJ). ... Clinical Features Fenichel (1978) and Engel (1980) reported patients with familial infantile or childhood myasthenic syndromes, noting that the syndromes can manifest as severe respiratory and feeding difficulties at birth. ... Among the 7 children with congenital myasthenic syndromes, severity varied. The diagnosis in severe cases was often obscured by apnea attacks, aspiration, and failure to thrive. ... Ohno et al. (2001) reported 5 patients, 4 males and 1 female, with a myasthenic syndrome associated with episodic apnea.
A number sign (#) is used with this entry because of evidence that presynaptic congenital myasthenic syndrome-21 (CMS21) is caused by homozygous or compound heterozygous mutation in the SLC18A3 gene (600336) on chromosome 10q11. ... Clinical Features O'Grady et al. (2016) reported 2 unrelated patients with congenital myasthenic syndrome apparent since infancy. The patients presented with hypotonia, apneas, and feeding difficulties in the first months of life.
A number sign (#) is used with this entry because of evidence that presynaptic congenital myasthenic syndrome-20 (CMS20) is caused by homozygous or compound heterozygous mutation in the SLC5A7 gene (608761) on chromosome 2q12. Description Congenital myasthenic syndrome-20 is an autosomal recessive neuromuscular disorder characterized by severe hypotonia associated with episodic apnea soon after birth.
A number sign (#) is used with this entry because presynaptic congenital myasthenic syndrome-24 (CMS24) is caused by homozygous or compound heterozygous mutation in the MYO9A gene (604875) on chromosome 15q23. ... INHERITANCE - Autosomal recessive HEAD & NECK Eyes - Ptosis - Nystagmus - Ophthalmoplegia - Oculomotor apraxia RESPIRATORY - Respiratory insufficiency - Respiratory crises during infection - Episodic apnea ABDOMEN Gastrointestinal - Swallowing difficulties - Poor feeding SKELETAL - Arthrogryposis, distal (in some patients) Limbs - Knee contractures Hands - Camptodactyly - Finger contractures Feet - Foot deformities MUSCLE, SOFT TISSUES - Myasthenic syndrome - Muscle weakness - Hypotonia, distal and proximal - Abnormal jitter seen on EMG - Decremental response to repetitive nerve stimulation NEUROLOGIC Central Nervous System - Delayed motor development - Delayed head control - Delayed walking - Impaired intellectual development, mild - Learning disabilities - Speech delay PRENATAL MANIFESTATIONS Movement - Decreased fetal movements MISCELLANEOUS - Onset in early infancy - Favorable response to treatment with 3,4-DAP or pyridostigmine MOLECULAR BASIS - Caused by mutation in the myosin IXA gene (MYO9A, 604875.0001 ) ▲ Close
A number sign (#) is used with this entry because of evidence that presynaptic congenital myasthenic syndrome-25 (CMS25) is caused by homozygous mutation in the VAMP1 gene (185880) on chromosome 12p13. Description Congenital myasthenic syndrome-25 is an autosomal recessive neuromuscular disorder characterized by hypotonia and generalized muscle weakness apparent from birth.
Mondor disease is a rare condition that is characterized by scarring and inflammation of the veins located just beneath the skin of the chest. The affected veins are initially red and tender and subsequently become a painless, tough, fibrous band that is accompanied by tension and retraction of the nearby skin. In most cases, the condition is benign and resolves on its own; however, Mondor disease can rarely be associated with breast cancer. Although the condition most commonly affects the chest, Mondor disease of other body parts (including the penis, groin, and abdomen) has been described, as well. Mondor disease is thought to occur when pressure or trauma on the veins causes blood to stagnate.
In many farsighted people, this vision problem is not part of a larger genetic syndrome. However, farsightedness (especially high hyperopia) can be a feature of other disorders with a genetic cause. Genetic conditions with farsightedness as a characteristic feature include microphthalmia, achromatopsia, aniridia, Leber congenital amaurosis, X-linked juvenile retinoschisis, Senior-Løken syndrome, Gorlin-Chaudhry-Moss syndrome, Down syndrome, and fragile X syndrome. ... When farsightedness is a feature of a genetic syndrome, it follows the inheritance pattern of that syndrome.
Forsius (1978) reported an 8-year-old boy with high-grade hyperopia and indicated that it is usually recessive. Forsius's colleague, Eriksson, has high hyperopia (Eriksson, 1978). Since his mother had the same refractive error, this may represent pseudodominance. Eyes - High-grade hyperopia Inheritance - Autosomal recessive vs. pseudodominance ▲ Close
A number sign (#) is used with this entry because of evidence that colobomatous macrophthalmia with microcornea (MACOM) is a contiguous gene deletion syndrome resulting from an approximately 22-kb heterozygous deletion on chromosome 2p22.2, involving the CRIM1 (606189) and FEZ2 (604826) genes. ... The sons of the proposita's sister presented an incomplete and unilateral pattern of these defects, suggesting variability in the expression of the syndrome. Pallotta et al. (1998) described a second family in which the syndrome was transmitted through 4 generations with no male-to-male transmission. ... In mice homozygous for loss of Crim1 in the head surface ectoderm and ocular mesenchyme, the authors observed morphologic changes overlapping the developmental eye anomalies present in patients with MACOM syndrome, including microcornea, a shallow anterior chamber, and a narrower eye without diminished axial diameter. Beleggia et al. (2015) concluded that CRIM1 is the causative gene for MACOM syndrome. No causative mutation or deletion/duplication involving CRIM1 was identified in the index patient from the family with MACOM described by Bateman and Maumenee (1984), suggesting further genetic heterogeneity of MACOM syndrome. INHERITANCE - Autosomal dominant HEAD & NECK Eyes - Nystagmus - Strabismus - Myopia - Reduced visual acuity - Microcornea - Moderate flattening of corneal curvature - Macrophthalmia - Pupillary dislocation - Coloboma of iris, choroid, and retina - Diffuse atrophy of retinal pigment epithelium - Macular atrophy - Strands of uveal tissue covering trabecular meshwork (in some patients) - Elevated intraocular pressure (in some patients) MISCELLANEOUS - Severity of reduced vision ranges from 20/50 to light perception only - Coloboma involves optic nerve in most patients MOLECULAR BASIS Contiguous gene syndrome caused by deletion of 22kb on 2p22.2 including the CRIM1 ( 606189 ) and FEZ2 ( 604826 ) genes ▲ Close
A rare genetic eye disease characterized by microcornea, coloboma of the iris and the optic disc, axial enlargement of the globe, staphyloma, and severe myopia. Additional manifestations are mild cornea plana, iridocorneal angle abnormalities with elevation of intraocular pressure, and shallow anterior chamber depth. Variable expressivity of the phenotype has been described, including unilateral or bilateral involvement, or variable extent of coloboma, among other features.
A number sign (#) is used with this entry because it represents a contiguous gene syndrome involving duplication of chromosome Xp11.23-p11.22. Clinical Features Giorda et al. (2009) described a syndrome characterized by borderline to severe mental retardation, speech delay, and EEG abnormalities associated with a microduplication at chromosome Xp11.23-p11.22. ... Cytogenetics Using array-based genomic hybridization to screen 2,400 individuals with isolated or syndromic mental retardation for copy number variation, Giorda et al. (2009) identified 8 (0.33%) unrelated individuals, 2 males and 6 females, with a microduplication at chromosome Xp11.23-p11.22. ... INHERITANCE - X-linked dominant GROWTH Weight - Excessive weight SKELETAL Feet - Pes cavus - Pes planus - Fifth toe hypoplasia - Syndactyly NEUROLOGIC Central Nervous System - Mental retardation, borderline to severe - Poor speech articulation - Subclinical absence seizures - EEG abnormalities - Diffuse paroxysmal discharges - Focal paroxysmal discharges - Continuous spike-wave discharges during sleep Behavioral Psychiatric Manifestations - Stubbornness - Shyness - Autistic-like features VOICE - Hoarse voice - Nasal voice ENDOCRINE FEATURES - Early puberty MISCELLANEOUS - Variable phenotype MOLECULAR BASIS - Contiguous gene syndrome associated with duplication (4.5 - 9.2Mb) of chromosome Xp11.22-p11.23 ▲ Close
Familial and de novo recurrent Xp11.22-p11.23 microduplication has been recently identified in males and females. Epidemiology To date, twelve patients have been described. Clinical description All patients show moderate to severe intellectual deficit and speech delay. Seizures, early puberty and lower-extremity anomalies, including pes planus or cavus, 5th toe hypoplasia, and syndactyly, are common. A peculiar electroencephalographic (EEG) pattern characterized by rolandic-like spikes and/or continuous spike wave during slow sleep (CSWS) exists in childhood. Etiology The microduplication was identified by microarray-based comparative genomic hybridization (aCGH).