-
Finnish Heritage Disease
Wikipedia
This has raised questions of bioethics and eugenics . [5] Contents 1 Finnish heritage disease types 2 Other genetic diseases 3 Genetic history 4 Etymology 5 See also 6 References Finnish heritage disease types There are 36 identified Finnish heritage diseases: [6] [7] Amyloidosis, Finnish type Lethal arthrogryposis with anterior horn cell disease Aspartylglucosaminuria Autoimmune polyendocrinopathy syndrome, type I , with or without reversible metaphyseal dysplasia Cartilage–hair hypoplasia Ceroid lipofuscinosis, neuronal , 1 Ceroid lipofuscinosis, neuronal, 3 Ceroid lipofuscinosis, neuronal , 5 Ceroid lipofuscinosis, neuronal, 8, Northern epilepsy variant (Synonyms: Northern epilepsy; Epilepsy, progressive, with mental retardation) Choroideremia Cohen syndrome Cornea plana 2 Diarrhea 1, secretory chloride, congenital Diastrophic dysplasia Epilepsy, progressive myoclonic 1A ( Unverricht–Lundborg ) Glycine encephalopathy (Nonketotic hyperglycinemia) GRACILE syndrome Gyrate atrophy of choroid and retina Hydrolethalus syndrome 1 Infantile-onset spinocerebellar ataxia ( Mitochondrial DNA depletion syndrome 7) Lactase deficiency , congenital Lethal congenital contracture syndrome 1 Lysinuric protein intolerance Meckel syndrome Megaloblastic anemia -1, Finnish and Norwegian type Mulibrey nanism Muscular dystrophy -dystroglycanopathy (congenital with brain and eye anomalies), type A, 3 Nephrotic syndrome, type 1 (Finnish congenital nephrosis) Ovarian dysgenesis 1 Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (Nasu–Hakola disease) Progressive encephalopathy with Edema, Hypsarrhythmia and Optic atrophy RAPADILINO syndrome Retinoschisis 1, X-linked, juvenile Sialuria, Finnish type (Salla disease) Tibial muscular dystrophy, tardive Usher syndrome , type 3A Out of these, three are rare causes of dwarfism : cartilage–hair hypoplasia , diastrophic dysplasia and Mulibrey nanism . Four genetically distinct subtypes of neuronal ceroid lipofuscinosis are found in the Finnish heritage: CLN1 , CLN3 , CLN5 , and CLN8 . [8] Names for conditions associated with these subtypes include infantile neuronal ceroid lipofuscinosis , Jansky–Bielschowsky disease and northern epilepsy syndrome . As of 2001, CLN5 and CLN8 had been reported almost exclusively in Finland. [8] Meckel syndrome type 1 ( MKS1 [9] ), a lethal condition, is known in 48 Finnish families. [10] Other genetic diseases The European Organization for Rare Diseases (EURORDIS) estimates that there are between 5,000 and 7,000 distinct rare diseases, affecting between 6% and 8% of the population of the European Union . [11] The majority of genetic diseases reported in Finland are not part of the Finnish disease heritage and their prevalence is not higher in Finland than worldwide. ... "Molecular diagnostics of Meckel–Gruber syndrome highlights phenotypic differences between MKS1 and MKS3". ... S2CID 11815792 . ^ Salonen R; Opitz, John M.; Reynolds, James F. (August 1984). "The Meckel syndrome: clinicopathological findings in 67 patients".
-
Kniest Dysplasia
Wikipedia
Alternative names for Kniest Dysplasia can include Kniest syndrome , swiss cheese cartilage syndrome , Kniest chondrodystrophy , or metatrophic dwarfism type II. ... v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteins
-
Pouchitis
Wikipedia
Contents 1 Signs and symptoms 2 Diagnosis 2.1 Classification 3 Treatment 4 Research 5 References 6 External links Signs and symptoms [ edit ] Symptoms of pouchitis include increased stool frequency, urgency, incontinence, nocturnal seepage, abdominal cramping, pelvic discomfort, and arthralgia . [4] Symptom severity does not always correlate with severity of endoscopically- or histologically-evaluated pouch inflammation. [4] Additionally, these symptoms are not necessarily specific for pouchitis, as they may arise from other inflammatory or functional pouch disorders such as Crohn's disease of the pouch, cuffitis , pouch sinus, or irritable pouch syndrome. [4] The most reliable tool for diagnosis is endoscopy combined with histologic features (derived from tissue biopsies obtained during endoscopy). [4] Diagnosis [ edit ] Classification [ edit ] Once a diagnosis of pouchitis is made, the condition is further classified. ... External links [ edit ] Classification D ICD - 10 : K91.8 ICD - 10-CM : K91.850 MeSH : D019449 External resources Orphanet : 217067 v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum v t e Inflammation Symptoms Flushing (Rubor) Fever (Calor) Swelling (Tumor) Pain (Dolor) Malaise Mechanism Acute Plasma-derived mediators Bradykinin complement C3 C5a MAC coagulation Factor XII Plasmin Thrombin Cell-derived mediators preformed: Lysosome granules biogenic amines Histamine Serotonin synthesized on demand: cytokines IFN-γ IL-8 TNF-α IL-1 eicosanoids Leukotriene B4 Prostaglandins Nitric oxide Kinins Chronic Macrophage Epithelioid cell Giant cell Granuloma Other Acute-phase reaction Vasodilation Increased vascular permeability Exudate Leukocyte extravasation Chemotaxis Tests Full blood count Leukocytosis C-reactive protein Erythrocyte sedimentation rate General Lymphadenopathy List of inflammed body part states
-
Diffuse Alveolar Damage
Wikipedia
Retrieved 2020-04-03 . ^ "Acute Respiratory Distress Syndrome (ARDS) | American Lung Association | American Lung Association" . www.lung.org . ... PMID 28828357 . ^ a b Agrawal, Rishi. "Respiratory distress syndrome | Radiology Reference Article | Radiopaedia.org" . ... "Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries" . ... "Diffuse alveolar damage associated mortality in selected acute respiratory distress syndrome patients with open lung biopsy" . ... PMID 16100134 . ^ a b Siegel, Mark D (March 2020). "Acute Respiratory Distress Syndrome: Prognosis and Outcomes in Adults" .CUX1, DCN, ASCC2, SARS2, SND1, TPX2, PSIP1, HPSE, SART3, PMEL, SFTPD, SFTPB, SDC1, SARS1, OAS3, NFKB2, MMP9, KRT14, IL17A, GTF2H4, FKBP4, H3P42

-
Tauopathy
Wikipedia
. ^ Kertesz A, McMonagle P, Jesso S (November 2011). "Extrapyramidal syndromes in frontotemporal degeneration". ... External links [ edit ] Classification D MeSH : D024801 v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteinsMAPT, GSK3B, CDK5, RMDN1, RMDN2, TARDBP, SNCA, RMDN3, SYBU, ALOX5, GABPA, UBB, NFE2L2, APOE, PRNP, REG1A, APP, PTPA, LRRK2, EIF2AK3, MSMB, RIDA, STXBP3, SIRT1, PSPH, MAPK8, BPIFA2, PSPN, HSP90AA1, TREM2, BDNF, MIR132, AHSA1, AIMP2, SGSM3, GRAP2, POLDIP2, SOD1, RNF19A, CRK, FKBP4, IGLON5, MAPK14, HSPA4, PIN1, PARK7, CX3CR1, MAPK1, ADNP, NPC1, HSPA8, OGT, HSPB2, SYNJ1, TP53, SYK, LMNA, IRS1, OGA, PSEN1, NECTIN3, COX2, PDC, MAPK9, SRSF2, TTBK1, GGT1, CASP3, STH, DLG4, EFHD2, GGTLC5P, GGT2, FUS, BIN1, AKT1, OPN1MW, GGTLC3, CTNNBL1, GGTLC4P, OPN1MW3, ACTB, OPN1MW2, GRN, RANBP9, DNM1L, MPHOSPH6, MIR219A1, KAT5, MIR155, AKT3, KHDRBS1, C9orf72, USP9X, HDAC6, SCRN1, NUAK1, USP2, PER2, CLOCK, MIR375, NPEPPS, SLC32A1, SQSTM1, USP13, NRXN3, HSPB3, LGI1, TTBK2, INPP5K, SLC26A7, NLRP3, GORASP1, PTBP2, CNTNAP2, MARK4, FGF21, RNF213, CD274, PSAT1, PRLH, SDF4, DCTN4, PPME1, VPS35, OTUB1, LCMT1, NMNAT1, NUP62, WNK1, EIF2A, FOXP2, TESC, HSPH1, CYP46A1, PPARGC1A, PHF6, SNW1, BACE1, NMNAT2, DNAJC5, PLEKHM2, SPHKAP, ZC3H14, DUSP26, PSCA, SERPINA3, TFEB, DDC, RCAN1, DYRK1A, MARK2, PTK2B, FAT1, FKBP5, MTOR, FYN, GABRG2, GFAP, GH1, GTF2H1, HTT, HDAC2, HMOX1, HSF1, HSPA1A, HSPA1B, HSPB1, IAPP, IGF1, DPP4, DCTN1, WFS1, CTSS, ADORA2A, GRK2, AIF1, APOB, KLK3, ARG1, ARNTL, BRCA1, TSPO, C3, C3AR1, CAPN2, CAST, CASP2, CD36, CETN1, CHI3L1, CREB1, CSF1R, CST3, CTSD, IGFALS, IL1A, IL1B, IL6, ROS1, RPS6KB1, SET, SFPQ, ACHE, TRA2B, SLC12A3, SMARCA1, SOAT1, SP1, SRPK2, SYT1, TH, TIA1, TLR4, TNF, TPO, TSC1, TTR, TYROBP, EZR, RELA, RBM4, PVALB, NGFR, IL10, LEP, MAP2, MECP2, MFGE8, MID1, MPZ, NEFL, NGF, NOS1, PTGS2, NR4A2, PRKN, PLA2G4A, PLAG1, PLP1, PRKAA1, PRKAA2, PRKAB1, PROS1, MTCO2P12

-
Multiple Endocrine Neoplasia Type 2b
Wikipedia
It was first described by Wagenmann in 1922, [3] and was first recognized as a syndrome in 1965-1966 by E.D. Williams and D.J. ... A variety of eponyms have been proposed for MEN 2B, such as Williams-Pollock syndrome, Gorlin-Vickers syndrome, and Wagenmann-Froboese syndrome. ... "Multiple mucosal neuromata with endocrine tumours: a syndrome allied to von Recklinghausen's disease". ... CS1 maint: multiple names: authors list ( link ) ^ Fryns JP, Chrzanowska K (October 1988). "Mucosal neuromata syndrome (MEN type IIb (III))" . J. Med. ... "Multiple endocrine neoplasia type 2B (mucosal neuroma syndrome, Wagenmann-Froboese syndrome)" .

-
Aldh18a1-Related De Barsy Syndrome
Orphanet
A rare, genetic, neurometabolic disease characterized by prenatal and postnatal growth retardation, hypotonia, failure to thrive, large and late-closing fontanel, development delay, cutis laxa, joint laxity, progeroid appearance, and dysmorphic facial features. In addition, corneal opacities, cataracts, myopia, seizures, hyperreflexia and athetoid movements have also been associated.
-
Thin Ribs-Tubular Bones-Dysmorphism Syndrome
Orphanet
An extremely rare, lethal, primary bone dysplasia characterized by thin ribs, thin long bones, high-arched palate and facial features of frontal bossing and low-set, posteriorly rotated ears. Bilateral cryptorchidism may be also observed. There have been no further descriptions in the literature since 1990.
-
Respiratory Bronchiolitis-Interstitial Lung Disease Syndrome
Orphanet
Respiratory bronchiolitis - interstitial lung disease is a mild inflammatory pulmonary disorder developed by cigarette smokers and characterized by shortness of breath and cough, pulmonary function abnormalities of mixed restrictive and obstructive lung disease and high resolution CT scanning showing centrilobular micronodules, ground glass opacities and peribronchiolar thickening.
-
Spastic Paraplegia-Facial-Cutaneous Lesions Syndrome
Orphanet
A complex form of hereditary spastic paraplegia characterized by delays in motor development followed by a slowly progressive spastic paraplegia (affecting mainly lower extremities) associated with a desquamating facial rash with butterfly distribution (presenting at around two months of age) and dysarthria. There have been no further descriptions in the literature since 1982.
-
48,xyyy Syndrome
Orphanet
A rare Y chromosome number anomaly that affects only males and is characterized by mild-moderate developmental delay (especially speech), normal to mild intellectual disability, large, irregular teeth with poor enamel, tall stature and acne. Radioulnar synostosis and clinodactyly have also been associated. Boys generally present normal genitalia, while hypogonadism and infertility is frequently reported in adult males.
-
Childhood-Onset Basal Ganglia Degeneration Syndrome
Orphanet
A rare genetic neurodegenerative disease characterized by sudden onset of progressive motor deterioration and regression of developmental milestones. Manifestations include dystonia and muscle spasms, dysphagia, dysarthria, and eventually loss of speech and ambulation. Brain MRI shows predominantly striatal abnormalities. The disease is potentially associated with a fatal outcome.
- Autosomal Recessive Cerebellar Ataxia-Epilepsy-Intellectual Disability Syndrome Due To Tud Deficiency Orphanet
-
Lowry-Wood Syndrome
Orphanet
A rare disorder characterized by the association of epiphyseal dysplasia, short stature, microcephaly and, in the first reported cases, congenital nystagmus. So far, less than 10 cases have been described in the literature. Variable degrees of intellectual deficit have also been reported. Other occasional features include retinitis pigmentosa and coxa vara. Transmission appears to be autosomal recessive.
-
Autoimmune Lymphoproliferative Syndrome Due To Ctla4 Haploinsuffiency
Orphanet
A rare, primary immunodeficiency characterized by variable combination of enteropathy, hypogammaglobulinemia, recurrent respiratory infections, granulomatous lymphocytic interstitial lung disease, lymphocytic infiltration of non-lymphoid organs (intestine, lung, brain, bone marrow, kidney), autoimmune thrombocytopenia or neutropenia, autoimmune hemolytic anemia and lymphadenopathy.
-
Usher Syndrome Type Ii
Gene_reviews
Prevalence The prevalence of Usher syndrome in the general US population has been conservatively estimated at 4.4:100,000. ... Usher syndrome has been estimated to be responsible for 3%-6% of all childhood deafness and approximately 50% of all deaf-blindness. These estimates were made prior to 1989, when Möller et al [1989] subdivided Usher syndrome into USH1 and USH2, and USH3 had not yet been recognized. ... Viral infections, diabetic neuropathy, and syndromes involving mitochondrial defects (see Mitochondrial Disorders Overview) can all produce concurrent symptoms of hearing loss and retinal pigmentary changes that suggest Usher syndrome. ... Safety and Efficacy of NPI-001 Tablets for Retinitis Pigmentosa Associated with Usher Syndrome (SLO RP). This is an interventional, two-year, Phase I/II clinical trial to evaluate the safety and efficacy of NPI-001 tablets in individuals with RP associated with Usher syndrome.
-
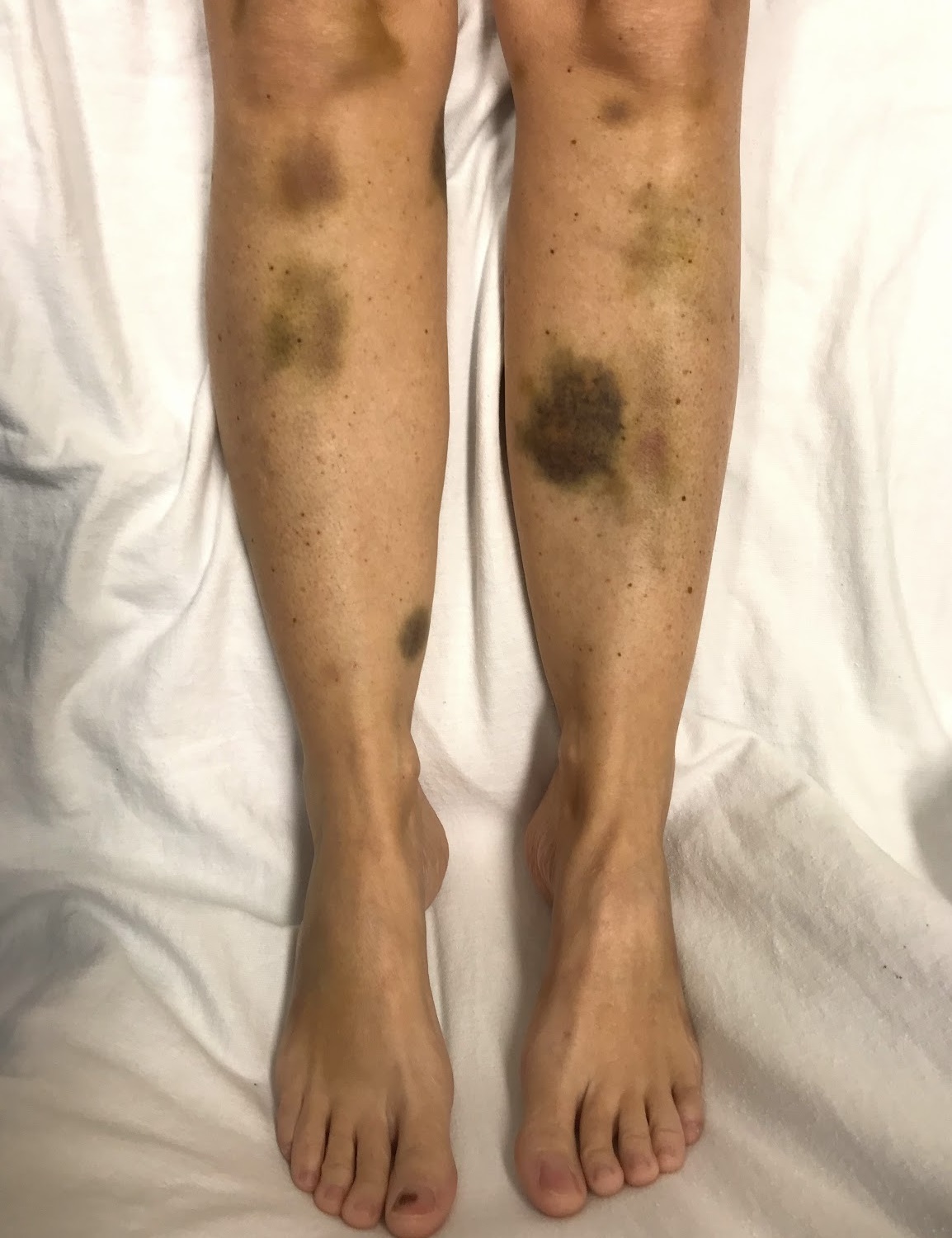
Thrombotic Thrombocytopenic Purpura
Wikipedia
Reportedly, less than 1% of all TTP cases are due to Upshaw–Schulman syndrome. [19] People with this syndrome generally have 5–10% of normal ADAMTS-13 activity. [18] [20] Secondary [ edit ] Secondary TTP is diagnosed when the person's history mentions one of the known features associated with TTP. ... This characteristic is shared by two related syndromes, hemolytic-uremic syndrome (HUS) and atypical hemolytic uremic syndrome (aHUS). [4] Consequently, differential diagnosis of these TMA-causing diseases is essential. ... ISBN 978-1560535164 . ^ a b c d Shatzel, JJ; Taylor, JA (March 2017). "Syndromes of Thrombotic Microangiopathy". ... "Heterogeneity of atypical haemolytic uraemic syndromes" . Arch. Dis. Child . 76 (6): 518–21. doi : 10.1136/adc.76.6.518 . ... "Interventions for haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura" .ADAMTS13, TFPI, F3, THBD, CFH, VWF, HLA-DRB1, PLG, SH3BP4, FLT4, ZFP36, HLA-DQB1, RBM45, CASP1, ABO, CRISP2, THBS1, DGKE, TNFSF10, CFLAR, PKD2L1, MAPKAPK2, CFHR3, TGFB1, WAS, ABCA1, PLA2G15, CCL2, MIR133B, PRSS55, RMDN2, KCNH8, TNFRSF13C, IL33, ACCS, SPZ1, CD248, ACSS2, ADAMTSL4, KRT20, TLR9, CLEC1B, PTPN22, STAT3, SERPINF2, S100B, MS4A1, EPHB1, ELAVL1, SLC25A10, CSF3, CRP, CD27, CD19, GHRH, CASP8, CASP3, CACNA1S, C3, APOH, APOA1, F2, BRF1, PTX3, CD46, PTGS2, PHEX, SERPINF1, SERPINE1, NRAS, COX2, JAK2, CFHR1, IL1B, IFNG, IFNB1, HP, HLA-DRB4, HLA-DRB3, MTCO2P12
-
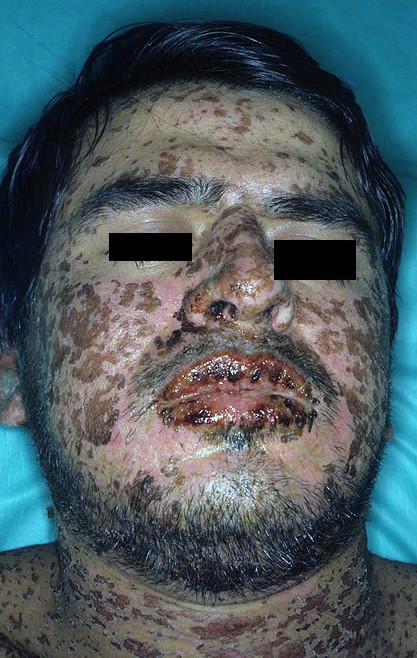
Stevens–johnson Syndrome
Wikipedia
"Current Perspectives on Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis". ... "The current understanding of Stevens–Johnson syndrome and toxic epidermal necrolysis" . ... "Nevirapine and the risk of Stevens–Johnson syndrome or toxic epidermal necrolysis". ... "Acetaminophen induced Steven Johnson syndrome-Toxic Epidermal Necrolysis overlap" . ... (November 2013). "Stevens–Johnson Syndrome (SJS) and Toxic Epidermal Necrolysis (TEN)" .HLA-B, HLA-A, HLA-C, IFNG, PTGER3, NOS2, IFNA2, MIF, NEDD4, NUCB1, ALB, EP300, PML, PSMC5, PTGIS, RB1, PARP1, RBX1, CAV1, DERL1, FBXO6, COPS5, CELF2, CTNNB1, VCP, ELMO1, CUL1, CUL4A, CSF3, NFKBIZ, POU5F1, TCF19, PSORS1C1, UMAD1, PSORS1C3, DDX39B, ATP6V1G2-DDX39B, GNLY, HSPG2, HLA-DRB1, TNF, TLR3, IKZF1, CYP2C9, CASP8, RBM45, TP53, CCL27, EGFR, MPRIP, CCL17, CYP2B6, CYP2C19, GZMB, RIPK3, HCP5, FAS, HLA-DQB1, ANXA1, CD33, ABCG2, SPAG9, WASF1, PROM1, CD37, CRP, SLC17A5, MIR18A, BAG6, MIR214, ZNF35, ALOX5, AKR1B1, UPP1, MICA, KEAP1, CASP9, FASLG, C5AR1, PART1, ARIH1, CABIN1, CD274, SETX, MARCHF1, PLXNA3, SCO2, TRAF3IP2, APC, PANK2, ABCB6, MLKL, BCL2L10, GSTM1, THBS1, TNFRSF1B, TLR2, COX2, CXCL9, FDXR, MDM2, LMNA, JAK2, IL15, IL13, IL10, IL4R, IL4, FPR1, IFNA13, FSHB, IFNA1, HMGB1, GFER, GJA1, CCR10, CFH, HDLBP, ETS2, ERCC1, NOS3, RET, TGFB2, TGFB1, SPTBN2, SOD1, SGSH, SFRP1, SDHD, SCT, SCN1A, CYP2C18, NPY, CYP2D6, CYP3A4, TIMM8A, DSP, EPHX1, ABCB1, PCM1, PAEP, ERBB2, PLG
-
Cardiomyopathy-Cataract-Hip Spine Disease Syndrome
Orphanet
Cardiomyopathy - cataract - hip spine disease describes the extremely rare triad of dilated cardiomyopathy, premature cataract, and articular disease of the hips and spine characterized by hip joint degeneration, irregular intervertebral disks, and platyspondyly. The ocular abnormalities are often the first symptoms to arise. There have been no further descriptions in the literature since 1985.
-
Absent Thumb-Short Stature-Immunodeficiency Syndrome
Orphanet
An exceedingly rare, autosomal recessive immune disease characterized by thumb aplasia, short stature with skeletal abnormalities, and combined immunodeficiency described in three sibships from two possibly related families. The skeletal abnormalities included unfused olecranon and the immunodeficiency manifested with severe chickenpox and chronic candidiasis. No new cases have been reported since 1978.




