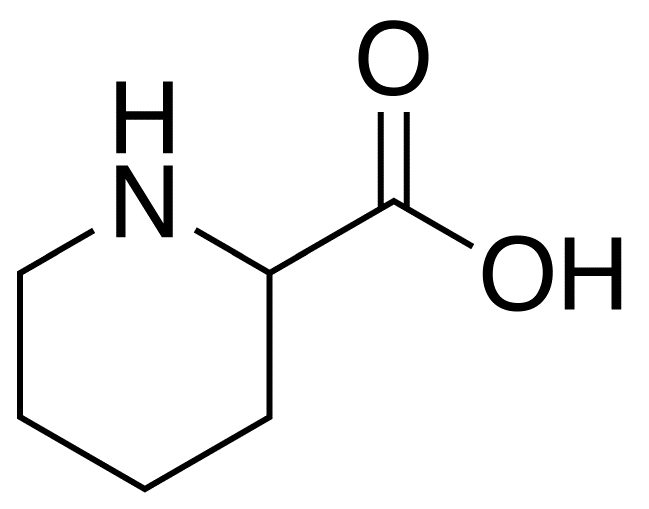
A number sign (#) is used with this entry because it represents a contiguous gene triplication syndrome (chr4:157.35-161.61). Clinical Features Wang et al. (2009) reported a mother and her 3 sons who all had a 4.3-Mb triplication of chromosome 4q32.1-q32.2 associated with delayed psychomotor development and variable mental retardation. ... INHERITANCE - Autosomal dominant HEAD & NECK Head - Macrocephaly Face - Frontal bossing - Long midface - Microretrognathia - Short philtrum - Hypoplastic zygoma Ears - Low-set ears - Small ears - Malformed ears - Squared-off ears Eyes - Downslanting palpebral fissures - Small palpebral fissures - Epicanthal folds - Ptosis Nose - Wide nasal bridge - Short nose ABDOMEN Gastrointestinal - Hirschsprung disease - Constipation NEUROLOGIC Central Nervous System - Hydrocephalus - Delayed psychomotor development - Speech abnormalities Behavioral Psychiatric Manifestations - Autistic features MISCELLANEOUS - One family has been reported (as of October 2010) MOLECULAR BASIS - Contiguous gene syndrome caused by 4.3-Mb triplication of chromosome 4q32.1-q32.2 ▲ Close
Summary Clinical characteristics. Hepatoerythropoietic porphyria (HEP) is characterized by blistering skin lesions, hypertrichosis, and scarring over the affected skin areas. Disease manifestations occur during infancy or childhood and with similar frequency in females and males. Individuals with HEP are not reported to be at increased risk for hepatocellular carcinoma. Diagnosis/testing. The diagnosis of HEP is established in a proband by identification of elevated porphyrins in the urine (predominantly uroporphyrin and heptacarboxylporphyrin) and significantly increased erythrocyte zinc protoporphyrin. Identification of biallelic pathogenic variants in UROD confirms the diagnosis.
Hepatoerythropioetic porphyria (HEP) is a very rare form of chronic hepatic porphyria (see this term) characterized by bullous photodermatitis. Epidemiology Fewer than 40 cases of HEP have been described. Clinical description The disease starts in childhood. The principle clinical signs include fragile skin, bullous cutaneous lesions that are sometimes erosive, and even mutilating on the surface of the skin exposed to the sun (hands, face). Hepatoerythropioetic porphria corresponds to homozygous and composite heterozygous cases of porphyria cutanea tarda (see this term). Etiology It is caused by a deficiency of uroporphyrinogen decarboxylase (URO-D; the fifth enzyme in the heme biosynthesis pathway) that leads to an accumulation of uroporphrin in the liver.
Hepatoerythropoietic porphyria (HEP) affects the skin and is due to a build-up of damaging chemicals in the body. Symptoms usually begin in infancy and include extreme sun sensitivity, extra body hair, discolored teeth, and anemia. Over time, people with HEP may lose skin, bone or develop scarring in sun-exposed areas. HEP is caused by a UROD gene that is not working correctly and is inherited in an autosomal recessive pattern. HEP is diagnosed based on the symptoms, clinical exam, laboratory testing, and confirmed by genetic testing.
The more severe type is known as hemoglobin Bart hydrops fetalis syndrome, which is also called Hb Bart syndrome or alpha thalassemia major. The milder form is called HbH disease. Hb Bart syndrome is characterized by hydrops fetalis, a condition in which excess fluid builds up in the body before birth. ... Thousands of infants with Hb Bart syndrome and HbH disease are born each year, particularly in Southeast Asia. ... The different types of alpha thalassemia result from the loss of some or all of these alleles. Hb Bart syndrome, the most severe form of alpha thalassemia, results from the loss of all four alpha-globin alleles. ... If both parents are missing at least one alpha-globin allele, their children are at risk of having Hb Bart syndrome, HbH disease, or alpha thalassemia trait.
Hemoglobin H (HbH) disease is a moderate to severe form of alpha-thalassemia (see this term) characterized by pronounced microcytic hypochromic hemolytic anemia. Epidemiology HbH disease is predominantly seen in Southeast Asia, the Middle East and the Mediterranean. Exact prevalence is not known but has been reported to be about 1/1,000,000 for severe forms of alpha-thalassemia in Northern Europe and North America. Clinical description Clinical features are highly variable and generally develop in the first years of life but may not develop until adulthood in some patients. Initial signs may be noticed only during routine hematologic analyses.
A rare form called alpha-thalassemia-intellectual deficit syndrome linked to chromosome 16 (16p13.3) has also been identified (see these terms). ... Alpha-thalassemia-intellectual deficit syndrome is characterized by very mild to severe anemia associated with developmental abnormalities. ... An acquired form known as alpha-thalassemia-myelodysplastic syndrome (ATMDS; see this term) has been described mainly in adult males and should also be considered.
Two types of alpha-thalassemia can cause health problems: the more severe type is known as Hb Bart syndrome; the milder form is called HbH disease. Hb Bart syndrome may be characterized by hydrops fetalis ; severe anemia; hepatosplenomegaly; heart defects; and abnormalities of the urinary system or genitalia. ... No treatment is effective for Hb Bart syndrome. For HbH disease, occasional red blood cell transfusions may be needed.
Alpha-thalassemia is usually inherited in an autosomal recessive manner. Hb Bart syndrome. At conception, each sib of a proband with Hb Bart syndrome has a 25% chance of having Hb Bart syndrome (e.g., --/--), a 50% chance of having α-thalassemia trait with deletion or inactivation of two α-globin genes in cis (e.g., --/αα), and a 25% chance of being unaffected and not a carrier. ... The severity of the α-thalassemia syndromes depends on the extent of α-globin chain defect (see Genotype-Phenotype Correlations). Hb Bart syndrome is the most severe clinical condition related to α-thalassemia. ... Hemoglobin Bart hydrops fetalis (Hb Bart) syndrome. See Prenatal Testing and Preimplantation Genetic Testing. ... Prevention of Primary Manifestations Hb Bart syndrome. Because of the severity of Hb Bart syndrome and the risk for maternal complications during pregnancy with a fetus with this disorder, prenatal diagnosis and early termination of affected pregnancies is usually considered.
A number sign (#) is used with this entry because Poretti-Boltshauser syndrome (PTBHS) is caused by homozygous or compound heterozygous mutation in the LAMA1 gene (150320) on chromosome 18p11. Description Poretti-Boltshauser syndrome is an autosomal recessive disorder characterized by cerebellar dysplasia, cerebellar vermis hypoplasia, cerebellar cysts in most patients, high myopia, variable retinal dystrophy, and eye movement abnormalities. ... The sibs in their twenties had normal IQ; 1 had Asperger syndrome. All 7 patients had eye movement abnormalities, including strabismus, oculomotor apraxia, amblyopia, and/or nystagmus, as well as myopia. ... Molecular Genetics In 7 patients from 5 unrelated families with Poretti-Boltshauser syndrome, Aldinger et al. (2014) identified homozygous or compound heterozygous mutations in the LAMA1 gene (see, e.g., 150320.0001-150320.0005).
A rare neuro-ophthalmological disease characterized by nonprogressive cerebellar ataxia, delayed motor and language development and intellectual disability, in addition to ophthalmological abnormalities (e.g. oculomotor apraxia, strabismus, amblyopia, retinal dystrophy and myopia). Cerebellar cysts, cerebellar dysplasia and cerebellar vermis hypoplasia, seen on magnetic resonance imaging, are also characteristic of the disease.
Clinical Features Smeitink and van den Heuvel (1999) described 2 brothers with complex I deficiency and features of Leigh syndrome (see 256000) confirmed postmortem. ... Lebon et al. (2007) reported 2 brothers, born of consanguineous Turkish parents, with complex I deficiency and Leigh syndrome resulting in death in infancy. ... Molecular Genetics In 2 brothers with complex I deficiency and features of Leigh syndrome confirmed postmortem, Smeitink and van den Heuvel (1999) identified a missense mutation in the NDUFS7 gene (V122M; 601825.0001). In a girl, born to consanguineous Tunisian parents, with severe complex I defect and Leigh syndrome, Lebon et al. (2007) identified homozygosity for a missense mutation in the NDUFS7 gene (R145H; 601825.0002). ... In 2 brothers, born of consanguineous Turkish parents, with complex I deficiency and Leigh syndrome, Lebon et al. (2007) identified a homozygous splice site mutation in the NDUFS7 gene (601825.0003).
Clinical Features Pagliarini et al. (2008) reported 2 Lebanese sibs, born of consanguineous parents, who presented in infancy with focal seizures, decreased movement and strength, ataxia, lactic acidosis, and neuroimaging results consistent with Leigh syndrome. Biochemical studies showed complex I deficiency in liver, muscle, and fibroblasts. ... Kohda et al. (2016) reported 4 unrelated children with mitochondrial complex I deficiency and Leigh syndrome. Clinical details were limited. Molecular Genetics In 2 Lebanese sibs, born of consanguineous parents, with mitochondrial complex I deficiency nuclear type 17 and Leigh syndrome, Pagliarini et al. (2008) identified a homozygous missense mutation in the NDUFAF6 gene (Q99R; 612392.0001) substitution in a highly conserved residue. ... In a 7-year-old boy, born of unrelated parents, with complex I deficiency and Leigh syndrome, Bianciardi et al. (2016) identified a heterozygous mutation in the NDUFAF6 gene (A178P; 612392.0002). ... Bianciardi et al. (2016) suggested that the second mutational event in the NDUFAF6 gene may be postmeiotic, affecting a nonexonic regulatory element and explaining the different tissue-specific expression, or that it may affect a specific protein In 4 unrelated children with mitochondrial complex I deficiency and Leigh syndrome, Kohda et al. (2016) identified biallelic missense mutations in the NDUFAF6 gene (612392.0003-612392.0007).
Overview Anal itching is a common condition. The itch in or around the anus is often intense and can be embarrassing and uncomfortable. Anal itching, also called pruritus ani (proo-RIE-tus A-nie), has several possible causes. They include infections, hemorrhoids and ongoing diarrhea. Skin inflammation, also called dermatitis, is another cause. If the symptoms don't clear up with self-care, talk with your health care provider. With treatment, most people get complete relief. Symptoms Symptoms of anal itching may include intense itching, inflammation, burning and soreness.
It mainly affects people with human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS). The cause of this condition isn't fully understood. ... If you have human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS), you may see improvement in your eosinophilic folliculitis symptoms after antiretroviral therapy.
A rare neurological disorder characterized by a reduced head circumference at birth with no gross anomalies of brain structure. It can be an isolated finding or it can be associated with seizures, developmental delay, intellectual disability, balance disturbances, hearing loss or vision problems.
This surgery relieves pressure on the brain, giving it enough space to grow and develop. Genetic changes. Down syndrome and other conditions may result in microcephaly.
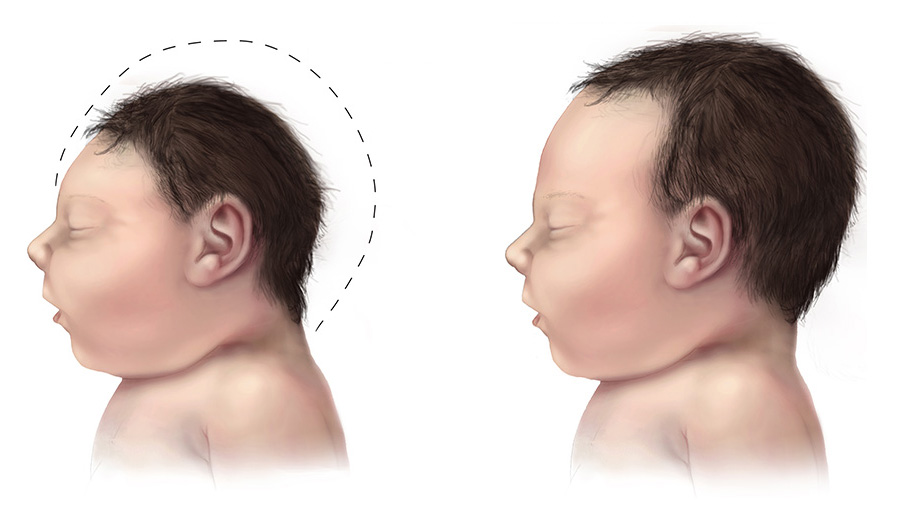
Microcephaly is a rare neurological condition in which a person's head is significantly smaller than expected based on standardized charts. Some cases of microcephaly are detected at birth, while others develop in the first few years of life. Some children with microcephaly have normal intelligence and development. However, microcephaly can be associated with seizures; developmental delay; intellectual disability; problems with movement and balance; feeding difficulties; hearing loss; and/or vision problems depending on the severity of the condition. Because the growth of the skull is determined by brain growth, the condition often occurs when the brain fails to grow at a normal rate.
A rare neurologic disease characterized by progressive sensorineural deafness, progressive sensory neuropathy and gastrointestinal abnormalities, including progressive loss of gastric motility and small bowel diverticulosis and ulcerations, resulting in cachexia. Additonal neurological manifestations may include dysarthria and absent tendon reflexes, as well as ptosis and external ophthalmoplegia. There have been no further descriptions in the literature since 1985.
A rare, genetic, dermis disorder characterized by bilateral, fairly symmetrical, antecubital webbing extending from distal third of humerus to proximal third of forearm, associated with musculoskeletal abnormalities (i.e. absent long head of triceps, bilateral posterior dislocation of the radial head and hypoplasia of the olecranon processes) and absent skin creases over the terminal interphalangeal joints of fingers, clinically manifesting with moderate to severe elbow extension and supination limitation.
Description Antecubital pterygium syndrome is an autosomal dominant disorder characterized by a fleshy web extending across the anterior aspect of the cubital fossa, absence of the long head of the triceps, limitation of full elbow extension, and missing skin creases over the terminal interphalangeal joints of the fingers (summary by Wallis et al., 1988). ... Inheritance The presence of affected persons of both sexes in 5 generations of the family reported by Wallis et al. (1988) indicated that the antecubital pterygium syndrome is inherited as an autosomal dominant trait.
Familial tumoral calcinosis (FTC) refers to a rare autosomal recessive disorder characterized by the occurrence of cutaneous and subcutaneous calcified masses, usually adjacent to large joints, such as hips, shoulders and elbows. FTC can occur in the setting of hyperphosphatemia or normophosphatemia, depending on the type of gene mutation involved.
A similar condition called hyperphosphatemia-hyperostosis syndrome (HHS) results in increased levels of phosphate in the blood, excessive bone growth, and bone lesions.
Summary Clinical characteristics. Hyperphosphatemic familial tumoral calcinosis (HFTC) is characterized by: Ectopic calcifications (tumoral calcinosis) typically found in periarticular soft tissues exposed to repetitive trauma or prolonged pressure (e.g., hips, elbows, and shoulders); and Painful swellings (referred to as hyperostosis) in the areas overlying the diaphyses of the tibiae (and less often the ulna, metacarpal bones, and radius). The dental phenotype unique to HFTC includes enamel hypoplasia, short and bulbous roots, obliteration of pulp chambers and canals, and pulp stones. Less common are large and small vessel calcifications that are often asymptomatic incidental findings on radiologic studies but can also cause peripheral vascular insufficiency (e.g., pain, cold extremities, and decreased peripheral pulses). Less frequently reported findings include testicular microlithiasis and angioid streaks of the retina. Diagnosis/testing. HFTC results from a relative deficiency of – or resistance to – the phosphate-regulating hormone, fibroblast growth factor 23 (FGF23).
The term 'hyperostosis-hyperphosphatemia syndrome' is sometimes used when the disorder is characterized by involvement of the long bones associated with the radiographic findings of periosteal reaction and cortical hyperostosis.
Grosse (1974) described associated cardiac, craniofacial and hand anomalies in a father and daughter. The cardiac defect was combined ventricular septal defect and pulmonic stenosis. The craniofacial 'defect' consisted mainly of narrow head and face. Very minor abnormalities were present in the hands, e.g., Dupuytren contractures in the father. Cardiac - Ventricular septal defect - Pulmonic stenosis Limbs - Minor hand anomalies - Dupuytren contractures Facies - Narrow head and face Inheritance - Autosomal dominant ▲ Close
A rare disorder of manganese transport characterized by childhood onset of extrapyramidal movement disorder (including dystonia, tremor, and bradykinesia), liver cirrhosis, polycythemia, and hypermanganesemia. Cases with spastic paraparesis without extrapyramidal dysfunction have also been reported. Cognitive functions are preserved. Brain imaging findings are consistent with deposition of manganese in the basal ganglia, dentate nucleus, brain stem, and anterior pituitary.
A number sign (#) is used with this entry because hypermanganesemia with dystonia-1 (HMNDYT1) is caused by homozygous mutation in the SLC30A10 gene (611146) on chromosome 1q41. Description Hypermanganesemia with dystonia-1 is an autosomal recessive metabolic disorder characterized by increased serum manganese, motor neurodegeneration with extrapyramidal features, polycythemia, and hepatic dysfunction, which leads to cirrhosis in some cases. Intellectual function is preserved (summary by Tuschl et al., 2012 and Quadri et al., 2012). Genetic Heterogeneity of Hypermanganesemia With Dystonia See also HMNDYT2 (617013), caused by mutation in the SLC39A14 gene (608736) on chromosome 8p21. Clinical Features Tuschl et al. (2008) reported an Arabic girl with a constellation of clinical features consisting of hypermanganesemia, liver cirrhosis, an extrapyramidal motor disorder, and polycythemia.
Hypermanganesemia with dystonia is an inherited disorder in which excessive amounts of the element manganese accumulate in the body (hypermanganesemia). One place manganese builds up in particular is in a region of the brain responsible for the coordination of movement, causing neurological problems that make controlling movement difficult. Consequently, the condition is characterized by involuntary, sustained muscle contractions (dystonia) and other uncontrolled movements. Two types of hypermanganesemia with dystonia, called hypermanganesemia with dystonia, polycythemia, and cirrhosis (HMDPC) and hypermanganesemia with dystonia 2, have been identified. They are distinguished by their genetic causes and certain specific features.
One patient had mucocutaneous pigment spots precisely like those of the Peutz-Jeghers syndrome. See blue rubber nevus syndrome (112200). Inheritance - Autosomal dominant GI - Cavernous hemangiomas of small intestine Skin - Mucocutaneous pigment spots as in Peutz-Jeghers syndrome ▲ Close
A rare ciliopathy characterized by the association of nephronophthisis and liver fibrosis. Renal manifestations include chronic renal failure, polyuria, polydipsia, anemia, as well as increased echogenicity on renal ultrasound and interstitial fibrosis and tubular dilation on biopsy. Hepatic involvement manifests as hepatosplenomegaly with extensive fibrosis, destruction of the bile ducts, and cholestasis. Mild psychomotor retardation and ocular symptoms, such as strabismus, nystagmus, retinal degeneration, and anisocoria, have been reported in some patients.
Mutations in the TMEM67 gene can also cause Meckel syndrome-3 (MKS3; 607361), Joubert syndrome-6 (JBTS6; 610688), and COACH syndrome (216360), all of which show more severe yet overlapping clinical features with NPHP.